A Non-Channel Function of CFTR: Attenuating Mitochondrial Oxidative Stress and Cardiomyocyte Senescence via Stabilization by USP45.
Cardiomyocyte senescence drives cardiovascular disease, underscoring the need to define its molecular mechanisms. The role of cystic fibrosis transmembrane conductance regulator (CFTR) ion channel in this process remains unclear, particularly regarding its expression and function. Atrial tissues were collected from patients with sinus rhythm or atrial fibrillation (AF) of varying durations. CFTR was downregulated in AF patients and negatively correlated with p16, p21, and p53. Myocardial aging models were established using D-galactose (D-gal) in both mice and neonatal mouse cardiomyocytes (CMs). In both animal and cellular models, D-gal increased SA-β-gal positivity and senescence markers while decreasing CFTR. Overexpressing CFTR reduced D-gal-induced elevations in p16, p21, p53, and malondialdehyde (MDA), and restored superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase (CAT) activities. Mechanistically, CFTR alleviates mitochondrial oxidative stress damage by enhancing plasma membrane Ca 2+ ATPase (PMCA) activity to reduce cytoplasmic Ca 2+ levels. Furthermore, we identified USP45 as a direct binding partner of CFTR, which deubiquitinates CFTR by specifically targeting K48-linked chains and the K688 residue. CFTR knockdown exacerbated D-gal-induced senescence and mitochondrial oxidative stress, which was rescued by USP45 overexpression. In conclusion, this study reveals a novel mechanism in which USP45-mediated deubiquitination of CFTR mitigates cardiomyocyte senescence and mitochondrial oxidative stress, offering a targeted intervention against age-related cardiovascular diseases.
Introduction
Cardiomyocyte senescence is a critical risk factor for various cardiovascular diseases, including myocardial infarction, cardiac fibrosis, arrhythmia, heart failure, and atherosclerosis (Song et al.2024). It can be triggered by multiple stressors such as DNA damage, oxidative stress, mitochondrial dysfunction, and epigenetic alterations (Zhang et al.2024). Senescent cardiomyocytes exhibit impaired contractility, increased pacing frequency, pronounced mitochondrial dysfunction, and hypertrophic growth, collectively contributing to cardiac dysfunction and remodeling (Duan et al.2023; Zhang et al.2024). Recent studies have demonstrated that genetic or pharmacological elimination of senescent cells holds potential for alleviating certain features of cardiac aging (Maggiorani et al.2024). Given the complexity of the mechanisms underlying cardiomyocyte senescence, understanding its key drivers and exploring targeted interventions are of substantial value for the development of novel therapeutic strategies for cardiovascular diseases.
The cystic fibrosis transmembrane conductance regulator (CFTR) is an adenosine triphosphate (ATP)‐gated anion channel that mediates the transport of Cl−and HCO3−, playing a crucial role in fluid transport and homeostasis (Liu et al.2024). Although CFTR is expressed in the heart, its cardiac functions remain incompletely understood (Wang et al.2020). Emerging evidence indicates that CFTR is involved in cardiomyocyte electrophysiological activity, and its silencing leads to impaired contractile function (Akita et al.2020; Sellers et al.2010). CFTR deficiency has been associated with defective cardiac development in zebrafish embryos and is linked to dilated cardiomyopathy (Liu et al.2020). Furthermore, dysregulated CFTR expression contributes to premature cellular senescence in murine lung tissue (Wellmerling et al.2019), and its reduced expression in skeletal muscle impairs autophagy and myogenesis, thereby exacerbating skeletal muscle aging (Chen et al.2025). However, the role of CFTR in cardiomyocyte senescence remains poorly investigated and warrants further exploration.
Mitochondria are involved in multiple interconnected cellular processes, including ATP production, nutrient metabolism, calcium homeostasis, and the regulation of programmed cell death (Ramaccini et al.2020). Mitochondrial dysfunction is a hallmark of cardiomyocyte senescence. In aging cardiomyocytes, the balance of mitochondrial fission and fusion is disrupted, leading to compromised functional integrity (Tang et al.2020). Accumulating evidence suggests that mitochondrial DNA mutations contribute to cardiovascular aging and related pathologies, while mitochondrial‐derived reactive oxygen species (ROS) may activate stress‐induced senescent phenotypes (Camacho‐Encina et al.2024; Stefanatos and Sanz2018). CFTR has been shown to confer protection against neuronal apoptosis following cerebral ischemia–reperfusion by mitigating mitochondrial oxidative stress (Zhang et al.2018). Furthermore, CFTR‐deficient mouse models and human lung epithelial cells exhibit reduced glutathione (GSH) levels and elevated oxidative stress (Artusi et al.2025). However, the role of CFTR in modulating mitochondrial oxidative stress in cardiomyocytes remains to be fully elucidated.
Ubiquitination plays a key role in regulating CFTR protein degradation and maintaining its peripheral quality control (Taniguchi et al.2023). Knockdown of STUB1 was shown to upregulate CFTR expression and alleviate calcium oxalate crystal‐induced renal tissue injury in mice (Hou et al.2024). In addition, ubiquitin protein ligase E3C (UBE3C) promotes the endoplasmic reticulum‐associated and peripheral degradation of misfolded CFTR (Kamada et al.2023). Okiyoneda et al. (2018) proposed that the peripheral protein quality control system can effectively recognize and ubiquitinate partially unfolded CFTR, underscoring the involvement of ubiquitination in the maintenance of CFTR stability. Shaw et al. (2010) demonstrated that serum/glucocorticoid‐regulated kinase 1 (SGK1)‐mediated phosphorylation of neural precursor cell expressed developmentally down‐regulated 4–2 (Nedd4‐2) inhibits its ubiquitination of CFTR, thereby increasing CFTR membrane abundance—highlighting the regulatory role of ubiquitination in modulating CFTR levels. However, whether deubiquitinating enzymes modulate mitochondrial function and cellular senescence phenotypes in cardiomyocytes via CFTR remains unclear.
This study is designed to identify ubiquitination‐related enzymes that regulate CFTR protein stability through a series of in vivo and in vitro experiments, and to further elucidate the roles and underlying mechanisms by which these enzymes and CFTR modulate mitochondrial function and senescence‐associated phenotypes in cardiomyocytes. Atrial fibrillation (AF) is closely associated with cardiomyocyte senescence, and during disease progression, it promotes adverse atrial remodeling (Adili et al.2022). The AF patient samples are used to indicate the clinical relevance of CFTR to myocardial senescence, and the subsequent mechanistic studies were conducted using an independent D‐galactose (D‐gal)‐induced senescence model, which does not involve direct investigation of arrhythmias. Our findings offer a theoretical foundation for developing novel therapeutic strategies against cardiovascular aging‐related diseases.
Methods and Materials
Clinical Data Collection
This study enrolled six patients with paroxysmal short‐term atrial fibrillation (SAF) and six with long‐standing permanent atrial fibrillation (LAF), all of whom underwent open‐heart surgery; six patients in sinus rhythm (SR) were included as controls. Patients older than 60 years of age (excluded to minimize age‐related confounding effects) or those with relevant comorbidities (e.g., malignant tumors or chronic inflammatory diseases) were not eligible for inclusion. Left atrial appendage (LAA) tissue samples were obtained from patients with AF and those in sinus rhythm during open‐heart surgery, rapidly frozen in liquid nitrogen, stored at −80°C, and subsequently used to analyze features of premature cellular aging in LAA tissue. All patients provided written informed consent and were admitted to Changsha Central Hospital. The study adhered to the principles outlined in the Declaration of Helsinki and was approved by the Ethics Committee of Changsha Central Hospital (2023‐151).
D‐Gal‐Induced Cardiac Aging Mouse Model
C57BL/6 male mice (6 months old, 20–30 g) were purchased from Hunan Silaike Jingda Laboratory Animal Company. The mice were housed under standard conditions: room temperature maintained at 22°C–25°C, relative humidity at 60% plus or minus 5%, and a 12‐h light/dark cycle. Food and water were freely available. After one week of acclimatization, mice were randomly divided into five groups (n= 5 per group): Sham group, D‐gal group, D‐gal+oe‐NC group, D‐gal+oe‐CFTR group, and D‐gal+oe‐USP45 group. Cardiac aging was induced by daily subcutaneous injection of D‐galactose (D‐gal, 120 mg/kg/day) into the posterior neck region for 12 weeks (Wang et al.2023). One week before modeling, mice in the D‐gal+oe‐NC and D‐gal+oe‐USP45 groups received slow tail vein injections of lentiviral vectors (1 × 109viral particles/mouse) (Zhao et al.2019). The USP45 overexpression lentiviral vector (oe‐USP45, Catalog number: HG‐LV027768) and the control vectors (oe‐NC) were purchased from Honorgene. After 12 weeks of modeling, transthoracic echocardiography was performed on mice under 3% isoflurane anesthesia using a Vevo 2100 high‐resolution imaging system (VisualSonics, FUJIFILM, Toronto, Canada) equipped with a 40.0 MHz phased‐array transducer. Cardiac function and structure were assessed based on changes in end‐systolic and end‐diastolic dimensions measured by M‐mode ultrasound. Ejection fraction (EF) and fractional shortening (FS) percentages, left ventricular anterior wall thickness at end‐systole (LVAW;s), left ventricular internal diameter at end‐systole (LVID;s), left ventricular posterior wall thickness at end‐systole (LVPW;s), left ventricular volume at end‐systole (LV Vol;s), left ventricular internal diameter at end‐diastole (LVID;d), left ventricular volume at end‐diastole (LV Vol;d), and left ventricular mass (LV Mass) were recorded. Following ultrasound examination, mice were euthanized by intraperitoneal injection of sodium pentobarbital (150 mg/kg). Cardiac tissues were collected for subsequent analysis. All animal experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committee of the South China University Affiliated Changsha Central Hospital (No. 2022‐S0031).
Hematoxylin and Eosin (H&E) and Masson Staining
Cardiac paraffin sections (4 μm) were deparaffinized and rehydrated. For H&E staining, sections were stained with hematoxylin (AWI0001a, Abiowell; 5 min) and eosin Y (G1100, Solarbio; 1 min), dehydrated through graded ethanol (95%–100%, 5 min/step), cleared in xylene, and mounted. For collagen detection, Masson's trichrome staining was performed per kit instructions (AWI0253a, Abiowell). Histopathology was assessed by light microscopy.
Neonatal Mouse Cardiomyocytes (NMCMs) Extraction and Processing
Newborn mice were anesthetized by inhalation of 3% isoflurane and then euthanized via cervical dislocation. NMCMs were isolated according to the following procedure (Liang et al.2015). Hearts were rapidly excised and transferred to a 35‐mm culture dish containing ice‐cold Hank's balanced salt solution (HBSS). The cardiac tissue was then minced into 1–3 mm3fragments and placed in a centrifuge tube with 5 mL of 0.08% trypsin. The tube was incubated in a 37°C water bath under gentle agitation for 6 min. After allowing the fragments to settle for 3 min, the supernatant was carefully aspirated. Next, 5 mL of fresh 0.125% collagenase II was added, and the tube was agitated again at 37°C for 15 min. The supernatant containing released cells was gently transferred to a new 15 mL centrifuge tube containing 5 mL of complete DMEM medium (supplemented with 10% FBS and 1% penicillin–streptomycin) to terminate trypsin activity. This digestion cycle was repeated 2–5 times until most tissue fragments were dissociated. The pooled cell suspension was centrifuged at 1000 rpm for 5 min. The pellet was resuspended in 6 mL of complete medium and seeded into a large culture dish. After 2 h of incubation, the unattached cells (enriched in cardiomyocytes) were collected from the supernatant, centrifuged, and resuspended in complete DMEM medium containing BrdU. This cell suspension was then seeded into culture flasks and maintained at 37°C in a 5% CO2incubator. Cells were passaged upon reaching confluence. The identity of NMCM was confirmed by immunofluorescence (IF) staining using an α‐actinin antibody.
NMCMs were treated with 40 μM D‐gal for 10 days to establish a cellular senescence model (Wang et al.2023). In the D‐gal+TEMPO group, NMCMs were exposed to 0.1 mM TEMPO (2,2,6,6‐Tetramethylpiperidin‐1‐oxyl, HY‐W001187, MCE), a mitochondrial ROS‐specific antioxidant, for 2 h following the 48‐h D‐gal stimulation [26]. Plasmid transfection was performed 48 h prior to the induction with D‐gal. Prior to D‐gal induction, NMCMs were pretreated with the proteasome inhibitors MG132 (10 μM) (Varney et al.2015) or carfilzomib (1 μM) (Halasi et al.2014) for 12 h, forming the D‐gal+MG132 and D‐gal+carfilzomib groups, respectively.
Cells Transfection
All plasmids used in this study were purchased from Honorgene. These include short hairpin RNA (shRNA) constructs for knocking down mouse CFTR (sh‐CFTR#1/2/3; Catalog: HG‐SH021050) and the plasma membrane Ca2+ATPase (PMCA) (sh‐PMCA#1/2/3; Catalog: HG‐SH1359507), overexpression plasmids for CFTR (oe‐CFTR; Catalog: HG‐MO021050) and USP45 (oe‐USP45; Catalog: HG‐MO027768), alongside their corresponding negative control vectors (sh‐NC and oe‐NC). Additionally, a series of ubiquitin mutant plasmids (K6, K11, K27, K29, K33, K48, K63; Catalogs: HG‐MO027768‐K6 to HG‐MO027768‐K63) and the K688R CFTR mutant plasmid (Catalog: HG‐MO021050‐K688) were also utilized. According to the manufacturer's protocol, plasmid transfection into NMCMs was performed using serum‐free medium and Lipofectamine 2000 (11668019, Invitrogen). After 6 h, the medium was replaced with fresh culture medium for continued incubation.
Senescence‐Associated β‐Galactosidase (SA‐β‐Gal) Staining
Mouse cardiac tissues were embedded in Optimal Cutting Temperature (OCT) compound and sectioned into 6‐μm‐thick frozen sections. Sections were immersed in distilled water to equilibrate to room temperature. SA‐β‐gal staining was performed using a β‐galactosidase staining kit (Cat# AWI0305a, Abiowell).
Briefly, sections were covered with 50 μL of SA‐β‐gal staining fixative solution and incubated at room temperature for 1 h. After washing with PBS, the staining working solution was applied to the sections. Sections were then incubated overnight at 37°C in a CO2‐free incubator. Following glycerol mounting, stained sections were examined and imaged under a light microscope (Model BA210T, Motic, China).
NMCMs were seeded in 6‐well plates. The cells were washed once with PBS and then fixed with 1 mL of β‐galactosidase staining fixative for 15 min. After three washes with PBS, 1 mL of staining working solution was added, and the plates were sealed with parafilm, followed by overnight incubation at 37°C. Staining results were observed under a microscope.
Western Blot (WB) Analysis
Mouse cardiac tissues or primary NMCMs were homogenized and lysed using RIPA lysis buffer. The protein concentration was determined with a BCA assay kit. Equal amounts of protein samples were separated by 10% SDS‐PAGE (75 V for 130 min) and subsequently transferred onto nitrocellulose (NC) membranes. The membranes were blocked with 5% skim milk for 90 min at room temperature, followed by incubation with specific primary antibodies overnight at 4°C. After washing, the membranes were incubated with the corresponding secondary antibodies for 90 min at room temperature. The protein bands were visualized using SuperECL Plus ultrasensitive luminescence solution (K‐12045‐D50, Advansta) and imaged with a ChemiScope 6100 imaging system (Clinx, China). Band intensity (gray value) was quantified using ImageJ software (National Institutes of Health, USA). GAPDH was used as the internal loading control. All antibody information is listed in Table1.
Table: Antibody information used in WB.
Biochemical Kit Test
According to the manufacturers' instructions, the levels/activities of malondialdehyde (MDA), superoxide dismutase (SOD), glutathione peroxidase (GSH‐Px), and catalase (CAT) in mouse serum, cardiac tissue, and/or NMCMs, as well as the levels of adenosine triphosphate (ATP), the nicotinamide adenine dinucleotide (NAD+)/reduced nicotinamide adenine dinucleotide (NADH) ratio, calcium ions (Ca2+), and chloride ions (Cl−) within NMCMs were measured using commercial assay kits. Except for the NAD+/NADH assay kit (S0175, Beyotime, China), all other kits were purchased from Nanjing Jiancheng Bioengineering Institute (China), with the following catalog numbers: MDA (A003‐1‐2), SOD (A001‐3‐2), GSH‐Px (A005‐1‐2), CAT (A007‐1‐1), ATP (A095‐1‐1), Ca2+(C004‐2‐1), and Cl−(C003‐2‐1).
Enzyme‐Linked Immunosorbent Assay (ELISA)
Additionally, ELISA kits were used to measure the secretion levels of senescence‐associated secretory phenotype (SASP) factors, including interleukin‐6 (IL‐6), interleukin‐8 (IL‐8), tumor necrosis factor‐alpha (TNF‐α), and C‐C motif chemokine ligand 2 (CCL2), in NMCMs and mouse serum. Kits for IL‐6 (KE10007), TNF‐α (KE10002), and CCL2 (KE10123) were from Proteintech, and the IL‐8 (CXCL15) kit (EM1592) was from Fine Test (https://fntest.cn/).
Transmission Electron Microscope (TEM)
Mouse cardiac tissue samples were fixed with glutaraldehyde and 1% osmium tetroxide, dehydrated through a graded series of acetone, and embedded in Epon‐812 resin. Ultrathin sections (70 nm) were mounted on copper grids and stained with uranyl acetate and lead citrate for 10 and 2 min, respectively. The sections were subsequently examined using a JEM‐1400FLASH transmission electron microscope (JEOL, Japan) to acquire images. For each group, myocardial tissue samples were obtained from 5 mice. From each sample, three non‐overlapping fields were randomly selected. Mitochondria were classified as damaged if they exhibited any of the following ultrastructural criteria: marked swelling, reduced matrix electron density, disrupted or absent cristae, vacuolization, or membrane discontinuity. Autophagosomes were identified as double‐membrane vacuoles containing cytoplasmic material/organelles. All analyses were performed in a double‐blind manner.
Flow Cytometry Analysis
Mitochondrial membrane potential (MMP) was assessed using a JC‐1 assay kit (C2003S, Beyotime). The JC‐1 working solution was prepared by diluting 50 μL of JC‐1 (200X) in 8 mL of ultrapure water, mixing thoroughly, and then adding JC‐1 Staining Buffer (5X). For JC‐1 staining of NMCMs, 600,000 cells were resuspended in 0.5 mL of cell culture medium. Then, 0.5 mL of the JC‐1 working solution was added to the cell suspension, followed by incubation at 37°C for 20 min in the dark. The cells were centrifuged at 600×gfor 3 min at 4°C, and the supernatant was carefully aspirated. The cell pellet was resuspended in 1 mL of JC‐1 Staining Buffer (1X) and centrifuged again at 600×gfor 5 min at 4°C. After discarding the supernatant, the cells were resuspended in an appropriate volume of JC‐1 Staining Buffer (1X). MMP was then immediately analyzed based on JC‐1 staining using a flow cytometer (A00‐1‐1102, Beckman). JC‐1 exists as a monomer in the cytoplasm, emitting green fluorescence (excitation/emission: 514/529 nm). When the mitochondrial membrane potential increases, JC‐1 forms aggregates that emit red fluorescence (excitation/emission: 585/590 nm). MMP was expressed as the ratio of red to green fluorescence intensity.
For the detection of mitochondrial reactive oxygen species (ROS), we utilized the MitoSOX Red reagent kit (S0061S, Beyotime, China). According to the manufacturer's protocol, NMCMs were incubated with 200 μL of the prepared MitoSOX Red working solution for 20 min at 37°C. After washing three times with serum‐free medium, the cells were resuspended and analyzed by flow cytometry.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT‐qPCR)
Total RNA was extracted from NMCMs using Trizol reagent. Messenger RNA (mRNA) was then reverse‐transcribed into complementary DNA (cDNA) using a commercial reverse transcription kit (CW2569, CWBIO, China). Quantitative real‐time polymerase chain reaction (RT‐qPCR) was performed on a QuantStudio 1 real‐time PCR system (Applied Biosystems, USA) using the UltraSYBR Mixture (CW2601, CWBIO, China). The thermal cycling conditions were as follows: initial pre‐denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension at 65°C for 30 s. The relative mRNA expression levels of the target genes were calculated using the 2−ΔΔCtmethod, with GAPDH serving as the internal reference gene. The primer sequences for all genes are listed in Table2.
Table: Primer sequences information.
Co‐Immunoprecipitation (CO‐IP) andIP
Protein was extracted from NMCMs using RIPA lysis buffer and aliquoted into Input, IP, and IgG tubes. The Input sample was processed normally for subsequent Western blot analysis. The IP and IgG tubes were incubated overnight at 4°C with a primary antibody against CFTR and control IgG antibody, respectively, followed by conjugation with Protein A/G agarose magnetic beads. After co‐immunoprecipitation, the samples were centrifuged at 3000 rpm for 3 min at 4°C. The resulting pellets were subjected to western blotting using antibodies against USP45 or ubiquitin (Ub).
Seahorse Assay
The mitochondrial oxygen consumption rate (OCR) of NMCMs was measured using an XFe96 Flux Analyzer (Seahorse Biosciences, Agilent). NMCMs (5 × 103cells/well) were seeded in XF‐96 plates with 80 μL DMEM and incubated overnight at 37°C. The mitochondrial stress test was performed by sequential addition of oligomycin (1.5 μM), carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP, 1.0 μM), and rotenone/antimycin A (each 0.5 μM). Hoechst dye was used for nuclear staining, and OCR values were normalized to cell count using a multi‐mode microplate reader (MB‐530, HEALES).
Cell Counting Kit‐8 (CCK‐8) Assay
NMCMs were seeded into 96‐well plates at a density of 5 × 103cells per well in 100 μL of DMEM and incubated at 37°C in a 5% CO2incubator until cell attachment. After the indicated treatments, 10 μL of CCK‐8 reagent (C0038, Beyotime, China) was added to each well, and the plates were further incubated for 4 h. The absorbance of each well was measured at 450 nm using a microplate reader.
5‐Ethynyl‐2′‐Deoxyuridine (EDU) Assay
NMCMs were inoculated at 5 × 103cells per well. Subsequently, cells were incubated with EDU (50 μM, C0078S, Beyotime, China) overnight at 37°C. After EDU incorporation, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X‐100. Nuclei were counterstained with Hoechst 33342. The percentage of EDU‐positive cells was quantified using a fluorescence microscope.
Apoptosis Detection
NMCMs were seeded into 96‐well plates at a density of 5 × 103cells per well in 100 μL of culture medium. After the indicated treatments, cells were digested with EDTA‐free trypsin and collected. The cells were then resuspended in 500 μL of Binding Buffer, followed by the addition of 5 μL of Annexin V‐APC (KGA1030, KeyGEN BioTECH, China) and 5 μL of propidium iodide (PI, MB2920, Meilun Bio, China). The mixture was gently vortexed and incubated for 10 min at room temperature in the dark. The apoptosis rate was immediately analyzed using a flow cytometer.
Data Analysis
Continuous data are reported as mean ± standard deviation. After verifying the assumptions of normality (using the Shapiro–Wilk test) and homogeneity of variances (using Bartlett's test), as appropriate, intergroup comparisons were made. Specifically, differences between two groups were analyzed by an unpaired Student'st‐test, while differences among more than two groups were analyzed by one‐way analysis of variance (ANOVA), followed by Tukey's honest significant difference test for post hoc comparisons. All statistical analyses were carried out with GraphPad Prism, version 8.0. Unless otherwise stated, a two‐tailedp‐value < 0.05 was considered to indicate statistical significance.
Results
The Aging of Myocardial Cells inAFPatients Is Accompanied by a Decrease inCFTRLevels
Aging is a major risk factor for AF, as senescence of atrial cardiomyocytes increases susceptibility to AF (Jansen et al.2025). SA‐β‐gal staining is currently the most commonly used and classic indicator for identifying cellular senescence. Several proteins associated with the senescence‐associated secretory phenotype (SASP) in cardiomyocyte senescence—including p16, p21, and p53 (Redgrave et al.2023). We assessed senescence markers in left atrial appendage (LAA) tissue obtained during open‐heart surgery from patients with normal sinus rhythm (Control group), short‐term atrial fibrillation (SAF), and long‐standing permanent atrial fibrillation (LAF). Compared to the Control group, expression of the senescence markers SA‐β‐gal (Figure1A), p16, p21, and p53 (Figure1B) was significantly elevated in the LAA tissue of both SAF and LAF patients. This increase was more pronounced in LAF patients than in SAF patients. Additionally, we found that cystic fibrosis transmembrane conductance regulator (CFTR) expression was reduced in the LAA tissue of AF patients, with LAF patients exhibiting lower CFTR levels than SAF patients (Figure1C). Pearson correlation analysis revealed a negative correlation between CFTR expression levels and p16, p21, and p53 levels in atrial tissue (Figure1D). These results indicate that the hearts of AF patients exhibit features of senescence, concomitant with downregulation of CFTR.
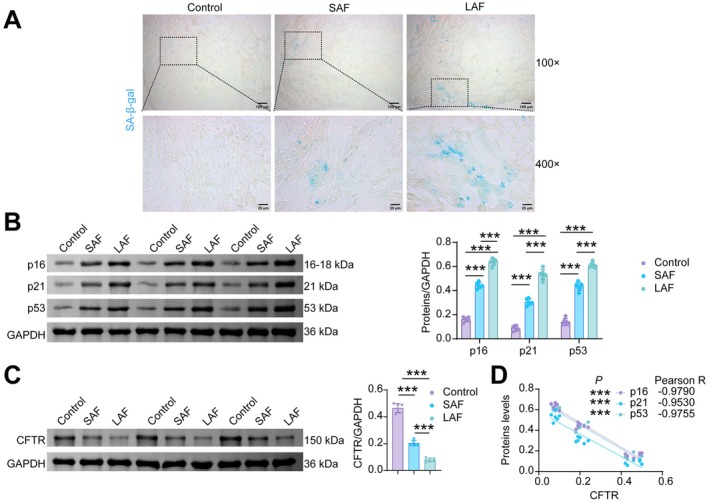
*Increased senescence markers and reduced CFTR expression in LAA of AF patients. (A) SA‐β‐gal staining. (B) WB analysis of p16, p21 and p53. (C) WB analysis of CFTR. (D) Pearson correlation analysis was performed to examine the correlations of CFTR with p16, p21, and p53 separately.n= 6, **p< 0.001.
CFTRReduction Occurs Alongside Mitochondrial Damage in Cardiomyocytes of D‐Galactose‐Induced Senescent Mice
Next, we validated CFTR expression in the D‐galactose (D‐gal)‐induced cardiac aging mouse model. In the Sham group, myocardial tissue exhibited compactly arranged cardiomyocytes with normal cellular architecture. Compared to the Sham group, D‐gal‐induced mice exhibited observable signs of cardiac remodeling, characterized by apparently enlarged cardiomyocyte nuclei (qualitative observation), myofiber disarray, and increased collagen deposition (Figure2A). Concurrently, SA‐β‐gal activity was elevated in the myocardial tissue of the model mice (Figure2B). Echocardiography revealed that D‐gal induction led to reduced ejection fraction (EF), fractional shortening (FS) percentages, left ventricular anterior wall thickness at end‐systole (LVAW;s), and left ventricular posterior wall thickness at end‐systole (LVPW;s), while left ventricular internal diameter at end‐systole (LVID;s), left ventricular internal diameter at end‐diastole (LVID;d), left ventricular volume at end‐diastole (LV Vol;d), and left ventricular mass (LV Mass) were increased (Figures2CandS1). Relative to the Sham group, model mice exhibited elevated malondialdehyde (MDA) levels in both serum (Figure2D) and myocardial tissue (Figure2E), accompanied by decreased activities of superoxide dismutase (SOD), glutathione peroxidase (GSH‐Px), and catalase (CAT). D‐gal treatment increased the protein expression of p16, p21, and p53 (Figure2F), as well as the levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in myocardial tissue (Figure2G), whereas it decreased CFTR expression (Figure2H). CFTR expression in mouse myocardium was negatively correlated with p16, p21, and p53 levels (Figure2I). These results demonstrate that CFTR is downregulated in D‐gal‐induced aging mouse hearts, concurrent with an increase in senescence markers.
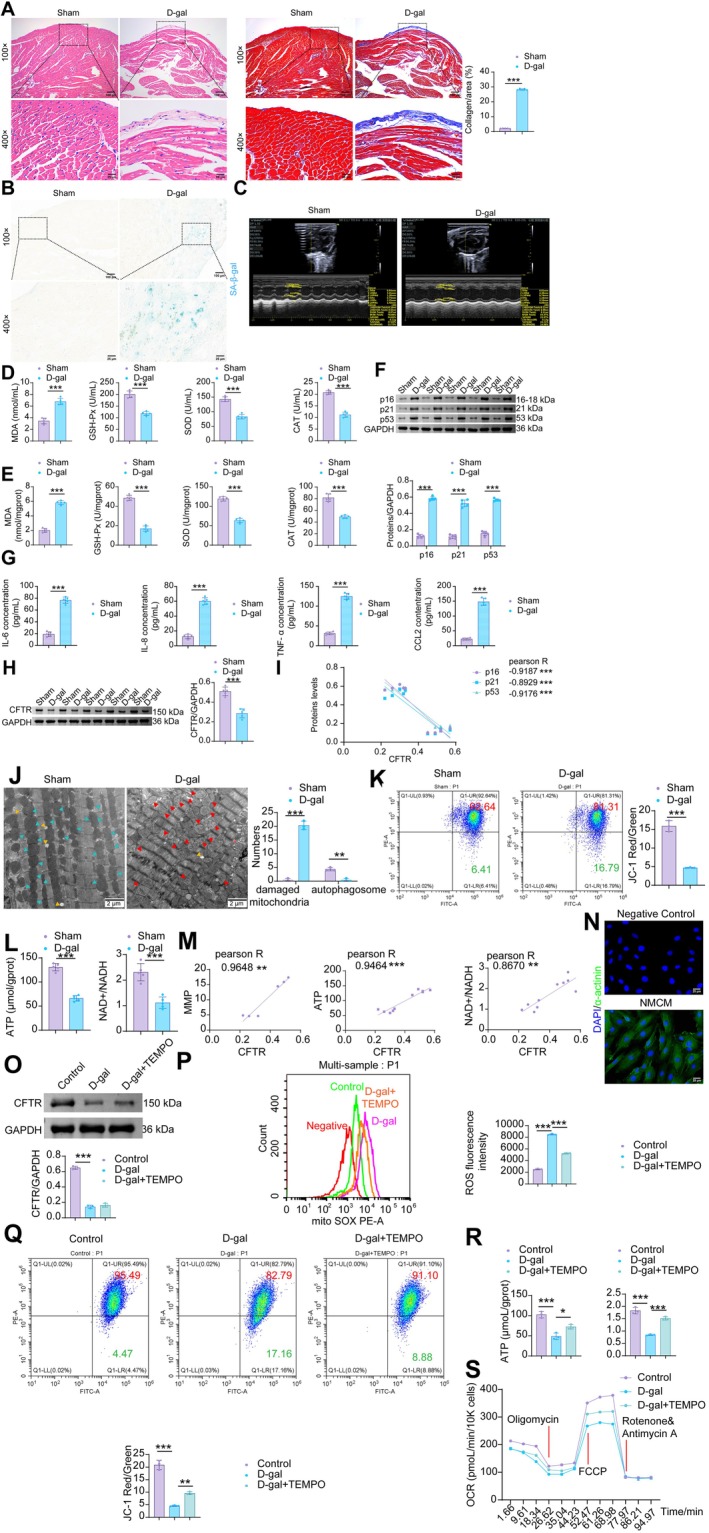
*The decrease in CFTR in cardiac myocytes coincides with elevated mitochondrial oxidative stress in the aging murine heart. (A) H&E and Masson staining of mouse myocardial tissue. (B) SA‐β‐gal staining in mouse myocardial tissue. (C) Echocardiography examination. (D) The levels of MDA, SOD, GSH‐Px and CAT in the peripheral blood of mice. (E) The levels of MDA, SOD, GSH‐Px and CAT in the mouse myocardial tissue. (F) The protein levels of p16, p21 and p53 in the mouse myocardial tissue. (G) The levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in the mouse myocardial tissue. (H) The protein levels of CFTR in the mouse myocardial tissue. (I) Pearson Correlation Analysis of CFTR with p53, p21, and p16 Expression. (J) Assessment of mitochondrial damage in mouse CMs by TEM. (K) Flow cytometric JC‐1 staining for the determination of MMP in mouse myocardial tissue. (L) ATP and NAD+/NADH levels in mouse myocardial tissue. (M) Pearson correlation analysis of CFTR with ATP and NAD+/NADH levels in mouse myocardial tissue. (O) IF staining of α‐actinin for extracting NMCMs. (N) WB analysis of CFTR in NMCMs. (P) Flow cytometric analysis of mitochondrial ROS in NMCMs. (Q) Flow cytometric JC‐1 staining for the determination of MMP in NMCMs. (R) ATP and NAD+/NADH levels in NMCMs. (S) Mitochondrial oxygen consumption rate (OCR) levels in NMCMs were measured using an XFe96 Flux Analyzer. Data are presented as mean ± SD. The sample sizes weren= 5 for Figure2A–Mandn= 3 for Figure2N–S. *p< 0.05, **p< 0.01, **p< 0.001.
Reportedly, aberrant CFTR expression may be associated with disrupted mitochondrial antioxidant defense (Rubin et al.2025). To further explore the relationship between CFTR and mitochondrial oxidative stress in mouse myocardial tissue, we conducted the following experiments. TEM analysis revealed a substantial increase in damaged mitochondria and a reduction in autophagosomes in cardiomyocytes after D‐gal induction (Figure2J). Compared to the Sham group, the D‐gal group showed a decrease in MMP (Figure2K), as well as reduced levels of ATP and NAD+/NADH (Figure2L). Pearson correlation analysis demonstrated positive correlations between CFTR expression and MMP, ATP, and NAD+/NADH levels (Figure2M). Isolated neonatal mouse cardiomyocytes (NMCMs) were identified by positive α‐actinin immunofluorescence staining, confirming successful isolation and culture (Figure2N). To determine the optimal time course for D‐gal‐induced cardiomyocyte senescence, we treated cardiomyocytes with D‐gal for 0, 1, 2, 5, and 10 days. The results showed that compared with the control group, cardiomyocytes exhibited decreased cell viability (FigureS2A) and proliferation (FigureS2B), increased apoptosis (FigureS2C), and upregulation of senescence‐associated proteins (p16, p21, and p53) as early as day 2 (FigureS2D). These senescent phenotypes became progressively more pronounced with longer treatment duration and were most evident at day 10. These findings indicate that although 48 h (day 2) of D‐gal treatment is sufficient to induce significant senescent changes in cardiomyocytes, the effect continues to strengthen over time. Therefore, we selected day 10, when the senescent phenotype was most robust, as the D‐gal modeling duration for our formal experiments. Compared to the Control group, D‐gal‐induced NMCMs exhibited decreased CFTR protein expression, which was not affected by treatment with the mitochondrial ROS‐specific antioxidant TEMPO (2,2,6,6‐Tetramethylpiperidin‐1‐oxyl) (Figure2O). D‐gal induction resulted in increased ROS generation in NMCMs, which was partially attenuated by TEMPO (Figure2P). In contrast, the D‐gal‐induced reductions in mitochondrial membrane potential (MMP), adenosine triphosphate (ATP), and the nicotinamide adenine dinucleotide (NAD+)/reduced nicotinamide adenine dinucleotide (NADH) ratio were partially reversed by TEMPO treatment (Figure2Q,R). Additionally, we performed Seahorse analysis to assess cellular mitochondrial stress. Our findings showed that the oxygen consumption rate (OCR) in D‐gal‐induced NMCMs was significantly reduced under both basal respiration and maximal respiration conditions, while TEMPO partially restored the D‐gal‐impaired OCR (Figure2S). These in vivo and in vitro results suggest that decreased CFTR expression in the aging heart may be positively associated with enhanced mitochondrial oxidative stress in cardiomyocytes.
CFTRAlleviates the Cardiac Aging Phenotype Induced by D‐Gal in Mice
To investigate the role of CFTR in D‐galactose‐induced cardiac senescence, we examined the effect of CFTR overexpression on senescence‐related phenotypes in mouse myocardial tissue. Compared with the D‐gal+oe‐NC group, CFTR overexpression not only elevated CFTR levels in cardiac tissue but also alleviated D‐gal‐induced disorganization of myocardial fibers and excessive collagen deposition (Figure3A,B). Lentivirus‐mediated delivery of oe‐CFTR partially reversed the D‐gal‐induced increase in MDA and the decrease in SOD, GSH‐Px, and CAT activities in both peripheral blood and myocardial tissue (Figure3C,D). Furthermore, CFTR overexpression reduced the elevated levels of senescence‐associated markers, including SA‐β‐gal, p16, p21, and p53, in cardiac tissues of D‐gal‐treated mice (Figure3E,F). CFTR overexpression partially reversed the D‐gal‐induced elevation of SASP factors (IL‐6, IL‐8, TNF‐α, CCL2) in mouse serum (Figure3G). These results demonstrate that CFTR overexpression attenuates D‐gal‐induced cardiomyocyte senescence.
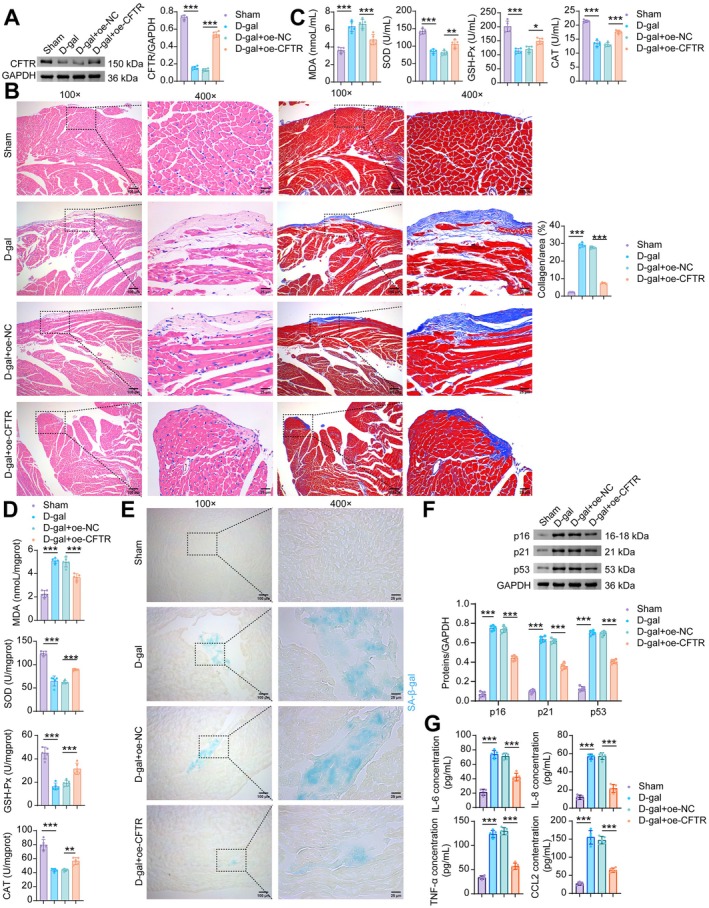
*Overexpression of CFTR rescues mice from D‐galactose‐induced cardiac senescence. (A) WB analysis of CFTR. (B) H&E and masson staining. (C) The levels of MDA, SOD, GSH‐Px and CAT in the serum of mice. (D) The levels of MDA, SOD, GSH‐Px and CAT in the mouse myocardial tissue. (E) SA‐β‐gal staining of the mouse myocardial tissue. (F) The levels of p16, p21, and p53 proteins in the mouse myocardial tissue. (G) The levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in the mouse myocardial tissue. Data are presented as mean ± SD,n= 5, *p< 0.05, **p< 0.01, **p< 0.001.
CFTRProtects Cardiomyocytes From D‐Gal‐Induced Oxidative Stress and Senescence
Deprivation of wild‐type CFTR in brain, heart, and lung tissues has been associated with inflammation and oxidative stress, and mutations in CFTR render mitochondria more susceptible to damage (Scialò et al.2024; Wu et al.2023). To further investigate the role of CFTR in D‐gal‐induced cardiomyocyte senescence and mitochondrial oxidative stress in vitro, we transfected cells with sh‐CFTR (#1, #2, #3), which resulted in reduced CFTR expression levels (Figure4A). The most effective knockdown construct, sh‐CFTR#3, was selected for subsequent experiments. Following D‐gal stimulation, CFTR expression was decreased in NMCMs. TEMPO, a superoxide dismutase mimetic, had no effect on CFTR expression. Compared with the D‐gal+TEMPO+sh‐NC group, transfection with sh‐CFTR further reduced CFTR expression in NMCMs (Figure4B). TEMPO treatment reduced the D‐gal‐induced increase in MDA levels in NMCMs, while CFTR knockdown partially counteracted this effect of TEMPO. The decreased levels of SOD, GSH‐Px, and CAT induced by D‐gal were elevated via TEMPO; however, CFTR knockdown partly reversed the protective effects of TEMPO (Figure4C). The positive rate of SA‐β‐gal staining was increased in D‐gal‐induced NMCMs. TEMPO reduced the increased SA‐β‐gal positive staining rate in D‐gal‐stimulated NMCMs. Inhibition of CFTR partially reversed the effect of TEMPO (Figure4D). Concurrently, CFTR knockdown elevated the expression of p16, p21, and p53, which were upregulated in D‐gal‐induced NMCMs but reduced by TEMPO (Figure4E). TEMPO treatment reduced the D‐gal‐induced secretion of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in NMCMs, whereas transfection with sh‐CFTR partially reversed the effects of TEMPO (Figure4F). Regarding mitochondrial function, the MMP of NMCMs decreased upon D‐gal stimulation. Compared to the D‐gal group, the addition of TEMPO increased the MMP of NMCMs, and this effect was attenuated by sh‐CFTR (Figure4G). Furthermore, TEMPO increased the reduced ATP and NAD+/NADH levels in D‐gal‐induced NMCMs, and these increases were reversed by CFTR knockdown (Figure4H). Notably, TEMPO treatment partially rescued the D‐gal‐reduced OCR, whereas knockdown of CFTR reversed these protective effects (Figure4I). These results suggest that CFTR may be involved in alleviating D‐gal‐induced cardiomyocyte senescence and mitochondrial oxidative stress.
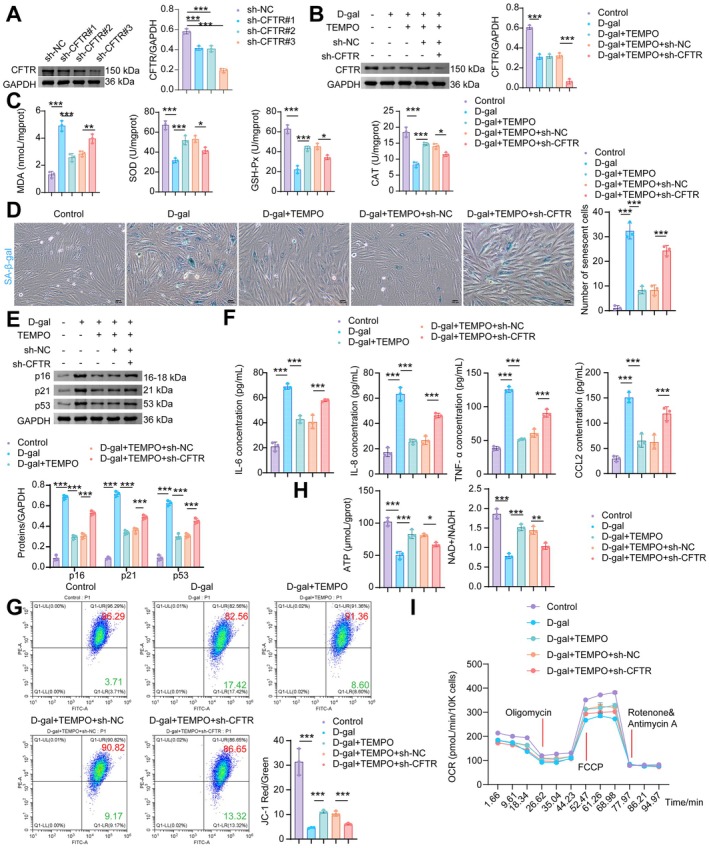
*CFTR inhibition aggravates D‐gal‐induced cardiomyocyte senescence and oxidative stress. (A, B) WB analysis of CFTR. (C) MDA, SOD, GSH‐Px, and CAT levels in NMCMs. (D) SA‐β‐gal staining. (E) WB analysis of p16, p21, and p53. (F) The levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in NMCMs. (G) MMP was assessed using JC‐1 staining followed by flow cytometry. (H) Levels of ATP, NAD+/NADH of NMCMs. (I) Mitochondrial OCR levels in NMCMs were measured using an XFe96 Flux Analyzer. Data are presented as mean ± SD,n= 3, *p< 0.05, **p< 0.01, **p< 0.001.
CFTRAlleviates Mitochondrial Oxidative Stress Damage by EnhancingPMCAActivity to Reduce Cytoplasmic Ca2+Levels
Previous research has demonstrated that reduced CFTR expression impairs PMCA function, leading to sustained cytosolic Ca2+elevation in ethanol‐treated mouse and human pancreatic organoids (Madácsy et al.2022). In NMCMs, overexpression of CFTR counteracted the D‐gal‐induced downregulation of PMCA expression (Figure5A). CFTR upregulation also reduced the D‐gal‐elevated levels of cytosolic Ca2+and Cl−(Figure5B,C). Next, we transfected D‐gal‐induced NMCMs with sh‐CFTR to examine the effect of CFTR knockdown on PMCA expression and its mediated calcium homeostasis. Transfection with sh‐CFTR further exacerbated the D‐gal‐induced downregulation of CFTR and PMCA protein expression in NMCMs (FigureS3A,B), as well as the elevations of intracellular Ca2+and Cl−(FigureS3C,D). Compared to the sh‐NC group, all sh‐PMCA constructs (#1, #2, #3) significantly knocked down PMCA expression (Figure5D). Among these, sh‐PMCA#3, which exhibited the most potent silencing effect, was selected for subsequent experiments. Transfection with oe‐CFTR in D‐gal‐treated NMCMs increased the expression of both CFTR and PMCA compared to the D‐gal+oe‐NC group (Figure5E) and reduced intracellular Ca2+accumulation (Figure5F). In contrast, compared to the D‐gal+oe‐CFTR+sh‐NC group, knockdown of PMCA did not affect CFTR expression but specifically reduced PMCA protein levels (Figure5E), and concurrently increased intracellular Ca2+(Figure5G). Furthermore, PMCA knockdown abolished the recovery of mitochondrial membrane potential (MMP) achieved by CFTR overexpression (Figure5H). CFTR overexpression in D‐gal‐treated NMCMs increased ATP production, the NAD+/NADH ratio (Figure5I), and OCR (Figure5J), downregulated the protein expression of senescence markers p16, p21, and p53 (Figure5K), decreased the percentage of SA‐β‐gal‐positive cells (Figure5L), and reduced the secretion of IL‐6, IL‐8, TNF‐α, and CCL2 (Figure5M). Importantly, inhibition of PMCA largely reversed these protective effects mediated by CFTR overexpression (Figure5G–M). These data indicate that CFTR mitigates mitochondrial impairment and cellular senescence via a PMCA‐mediated reduction in cardiomyocyte Ca2+accumulation.
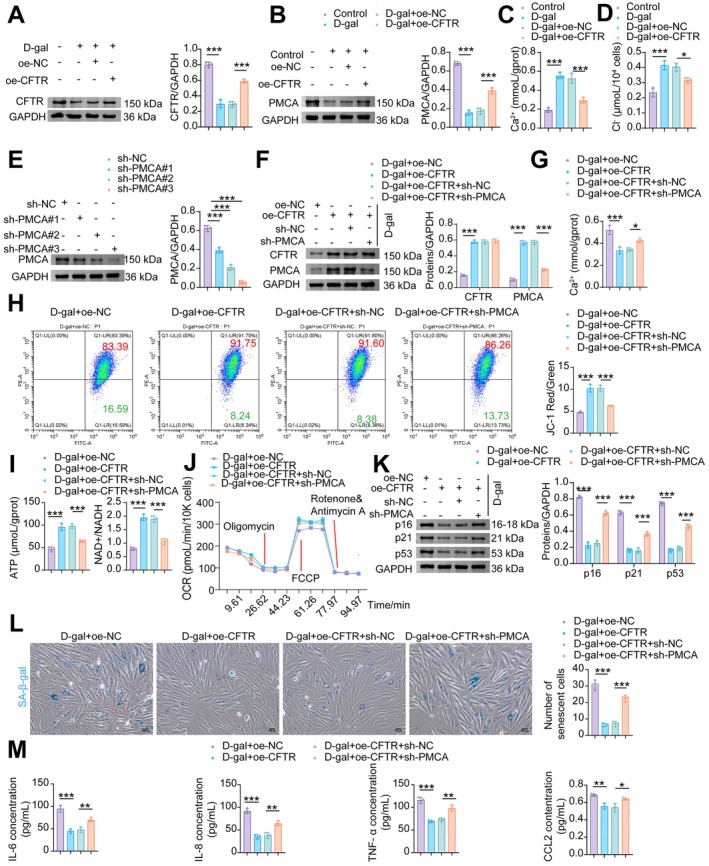
*PMCA‐mediated Ca2+ clearance is required for CFTR to ameliorate mitochondrial dysfunction and cellular senescence in D‐gal‐treated NMCMs. (A) WB analysis of PMCA. (B) Levels of Ca2+. (C) Levels of Cl−. (D) WB analysis of PMCA. (E) WB analysis of CFTR and PMCA. (F) Levels of Ca2+. (G) Flow cytometric analysis of MMP with JC‐1. (H) Levels of ATP and NAD+/NADH. (I) SA‐β‐gal staining was performed to detect senescent cells. (J) Mitochondrial OCR levels in NMCMs were measured using an XFe96 Flux Analyzer. (K) WB analysis of p16, p21, and p53. (L) SA‐β‐gal staining in NMCMs. (M) The levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in NMCMs. Data are presented as mean ± SD,n= 3, *p< 0.05, **p< 0.01, **p< 0.001.
USP45Prevents the Degradation ofCFTRby Inhibiting Its Ubiquitination
Inhibition of ubiquitination to stabilize the CFTR protein has long been recognized as a therapeutic target for various diseases, such as cystic fibrosis and kidney injury (Hou et al.2024; Okiyoneda et al.2024). We found that D‐galactose (D‐gal) and the proteasome inhibitors MG132 and carfilzomib did not affect CFTR mRNA expression (Figure6A). However, MG132 and carfilzomib increased CFTR protein expression, which was reduced by D‐gal (Figure6B), and attenuated D‐gal‐enhanced CFTR ubiquitination (Figure6C). Subsequently, we queried the UbiBrowser 2.0 database (http://ubibrowser.bio‐it.cn/) and identified known deubiquitinating enzymes of CFTR, including USP10, as well as predicted ones such as USP9X, USP33, and USP45 (Figure6D). Experimental validation showed that, compared with the control group, the expression of USP45 and USP9X was downregulated in D‐gal‐induced NMCMs, while the expression of USP10 and USP33 remained unchanged (Figure6E). Among these, USP45, which exhibited the most significant downregulation, was selected for further investigation. Co‐immunoprecipitation (Co‐IP) results revealed a specific interaction between CFTR and USP45 proteins (Figure6F). Under pathological conditions of aging, the downregulation of USP45 is a key factor leading to reduced CFTR protein levels in NMCMs.
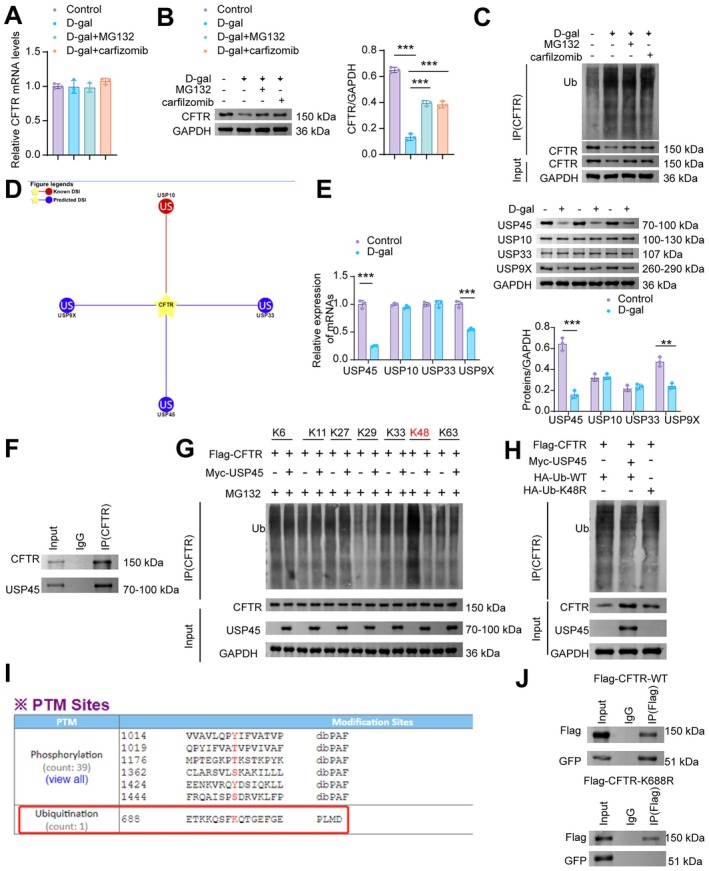
*CFTR is deubiquitinated by USP45 at the K688 residue and on K48‐linked ubiquitin chains. (A) RT‐qPCR analysis of CFTR mRNA. (B) WB analysis of CFTR. (C) D‐gal‐induced NMCMs were treated with MG132 (10 μM for 12 h) and carfilzomib (1 μM for 12 h), followed by immunoprecipitation (IP) to assess the ubiquitination level of CFTR. (D) Prediction of CFTR‐targeting deubiquitinating enzymes was performed using the UbiBrowser 2.0 database. (E) RT‐qPCR and WB analysis of USP45, USP10, USP33, USP9X. (F) CO‐IP of CFTR and USP45. (G) Identification of USP45‐targeted ubiquitin linkages on CFTR by IP/WB. (H) Overexpression of USP45 or mutation of the ubiquitin chain residue to K48R (K48R mutant) resulted in the suppression of CFTR ubiquitination. (I) The GPS‐PTMD database predicted K688 as a potential ubiquitination site on CFTR. (J) NMCMs were co‐transfected with GFP‐USP45 and either WT or K688R CFTR fragments for 48 h. Proteins were pulled down using IgG and an anti‐CFTR antibody, and further analyzed by WB with antibodies against CFTR and USP45 (GFP). Data are presented as mean ± SD,n= 3, **p< 0.01, **p< 0.001.
To elucidate the mechanism by which USP45 mediates CFTR deubiquitination, we examined the types of ubiquitin chains (e.g., K48‐linked, K63‐linked) attached to CFTR and the modification levels at its ubiquitination sites after USP45 overexpression. Overexpression of USP45 significantly reduced ubiquitination at the K48 site of CFTR (Figure6G). Compared with transfection of NMCMs with HA‐tagged wild‐type ubiquitin (HA‐Ub‐WT), both HA‐tagged K48‐only mutant ubiquitin (HA‐Ub‐K48R) and USP45 overexpression plasmids decreased CFTR ubiquitination levels and enhanced CFTR protein stability (Figure6H). The GPS‐PTMD database predicted K688 as a ubiquitination site of CFTR (http://ptmd.biocuckoo.org/finalpage.php?uniprot=P13569) (Figure6I). After successful transfection of cardiomyocytes with GFP‐tagged USP45, USP45 was unable to bind to the K688R CFTR mutant, unlike wild‐type CFTR (WT‐CFTR) (Figure6J). These results demonstrate that USP45 specifically targets the K688 site of CFTR and removes K48‐linked polyubiquitin chains, thereby counteracting CFTR degradation via the proteasome pathway and enhancing its stability.
USP45Alleviates Mitochondrial Oxidative Stress Damage and Cellular Senescence in Cardiomyocytes by Enhancing the Stability of theCFTRProtein
In vitro, we further investigated whether USP45 alleviates mitochondrial oxidative stress and cellular senescence in cardiomyocytes by regulating CFTR protein stability. Compared to the D‐gal+oe‐NC group, transfection with oe‐USP45 upregulated both USP45 and CFTR expression in D‐gal‐treated NMCMs. Knockdown of CFTR downregulated its own expression but did not affect USP45 levels in NMCMs (Figure7A). Overexpression of USP45 reduced intracellular Ca2+levels (Figure7B), restored the D‐gal‐induced loss of mitochondrial membrane potential (MMP) (Figure7C), and increased the levels of ATP and the NAD+/NADH ratio, which were impaired by D‐gal treatment (Figure7D). These beneficial effects were abolished by CFTR inhibition (Figure7B–D). While USP45 overexpression partially restored the D‐gal‐suppressed OCR, CFTR knockdown reversed the effect of oe‐USP45 compared with the D‐gal+oe‐USP45+sh‐NC group (Figure7E). Furthermore, USP45 overexpression reduced the proportion of senescent NMCMs induced by D‐gal (Figure7F) and decreased the protein expression of senescence markers p16, p21, and p53 (Figure7G). Knockdown of CFTR reversed the anti‐senescence effects of USP45 overexpression (Figure7F–G). Compared with the D‐gal+oe‐NC group, USP45 overexpression suppressed the secretion of IL‐6, IL‐8, TNF‐α, and CCL2 in NMCMs, whereas CFTR knockdown completely reversed the effect of oe‐USP45 (Figure7H). These results demonstrate that USP45 attenuates mitochondrial damage and cellular senescence in cardiomyocytes by upregulating CFTR protein stability.
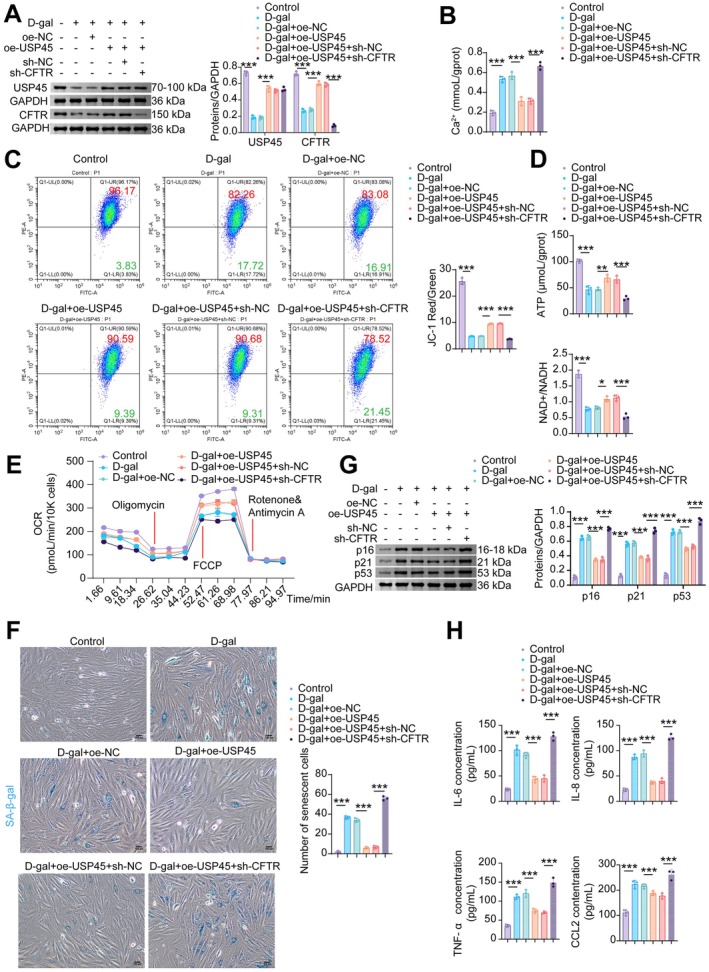
*USP45 attenuates mitochondrial damage and senescence in CMs by upregulating CFTR protein stability. (A) WB analysis of USP45 and CFTR. (B) The Ca2+level in NMCM. (C) MMP was measured by JC‐1 flow cytometry. (D) ATP and NAD+/NADH levels. (E) Mitochondrial OCR levels in NMCMs were measured using an XFe96 Flux Analyzer. (F) SA‐β‐gal staining. (G) WB analysis of p16, p21, and p53. (H) The levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in NMCMs. Data are presented as mean ± SD,n= 3, *p< 0.05, **p< 0.01, **p< 0.001.
USP45StabilizesCFTRto Attenuate Cardiac Aging and Oxidative Stress
In vivo, we further validated whether USP45 alleviates myocardial cell senescence and mitochondrial damage in mice via CFTR. Both USP45 and CFTR showed downregulation in D‐gal‐induced mice myocardial tissues. Compared with the D‐gal+oe‐NC group, USP45 overexpression reversed the reduced expression of both USP45 and CFTR in the myocardial tissues of D‐gal‐induced senescent mice (Figure8A). Myocardial fibers in the control group were compactly and neatly arranged with clear structure, normal intercellular space, regular nuclear morphology, and centrally located nuclei. In contrast, the D‐gal group exhibited disordered myocardial tissue structure, including partially broken, dissolved, or necrotic myocardial fibers; swollen cardiomyocytes; and pyknotic or fragmented nuclei in some cells. Additionally, extensive diffuse blue collagen fiber deposition and significant fibrosis were observed in the myocardial interstitium, with a significantly higher collagen volume fraction compared to the control group. In comparison with the D‐gal+oe‐NC group, the D‐gal+oe‐USP45 group showed significant improvement in myocardial histopathological injury: myocardial fibers were more regularly arranged with reduced disruption, and cardiomyocyte swelling and necrosis were alleviated. Nuclear morphology was also more regular. Masson's trichrome staining further confirmed that USP45 overexpression markedly attenuated the degree of myocardial interstitial fibrosis and reduced collagen deposition in model mice compared to the D‐gal+oe‐NC group (Figure8B). Echocardiography results demonstrated that USP45 overexpression partially reversed D‐gal‐induced alterations in cardiac function and ventricular structure. Specifically, it increased the D‐gal‐reduced EF, FS, LVAW;s, and LVPW;s, and decreased the D‐gal‐elevated LVID;s, LV Vol;s, LVID;d, LV Vol;d, and LV Mass (Figures8CandS4). Furthermore, USP45 overexpression elevated the reduced mitochondrial membrane potential in senescent model mice (Figure8D), decreased the D‐gal‐induced elevation of MDA levels in serum and myocardial tissue, and increased the D‐gal‐reduced levels of SOD, GSH‐Px, and CAT (Figure8E,F). The positive rate of SA‐β‐gal staining in myocardial tissues of senescent model mice was reduced by oe‐USP45 (Figure8G), and the protein expression of senescence markers p16, p21, and p53 was downregulated by USP45 overexpression (Figure8H). USP45 overexpression partially inhibited the secretion of SASP factors elevated by D‐gal (Figure8I). These results suggest that USP45 may alleviate mitochondrial damage and cellular senescence phenotypes in cardiomyocytes of aged mice by stabilizing the CFTR protein.
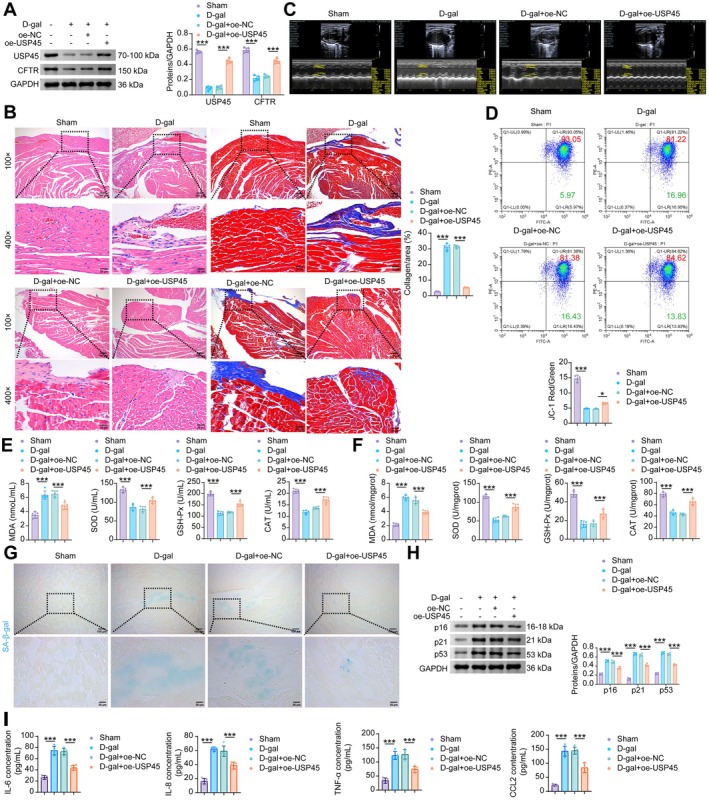
*USP45 rescues cardiac aging and mitochondrial damage by mediating CFTR stabilization. (A) WB analysisi of USP45 and CFTR in the mouse myocardial tissue. (B) H&E and Masson staining of mouse myocardial tissue. (C) Echocardiography examination. (D) Flow cytometric JC‐1 staining for the determination of MMP in mouse myocardial tissue. (E) The levels of MDA, SOD, GSH‐Px and CAT in the serum of mice. (F) The levels of MDA, SOD, GSH‐Px and CAT in the mouse myocardial tissue. (G) SA‐β‐gal staining in mouse myocardial tissue. (H) The protein levels of p16, p21 and p53 in the mouse myocardial tissue. (I) The levels of SASP factors (IL‐6, IL‐8, TNF‐α, and CCL2) in the mouse myocardial tissue. Data are presented as mean ± SD,n= 5, *p< 0.05, **p< 0.001.
Discussion
Although the effects of aging have been extensively studied, the process of cardiomyocyte senescence remains poorly understood. Studies have shown that mitochondrial function and energy transduction efficiency are both impaired during the senescence of cardiomyocytes (Tepp et al.2017). In the context of cardiac aging, alterations in metabolic substrates and enzyme activities lead to increased oxidative stress, elevated levels of ROS, and are accompanied by mitochondrial dysfunction and changes in gene expression (Hao and Liu2023). This study reveals that USP45 alleviates D‐gal‐induced myocardial senescence and mitochondrial oxidative stress in mice by modulating the ubiquitination of CFTR.
The prevalence of AF increases with advancing age, and cardiomyocyte senescence is more pronounced in the AF population (Adili et al.2022). This aligns with our findings, which indeed demonstrate more evident myocardial senescence in AF patients compared to healthy individuals. Furthermore, analysis of clinical samples in this study indicates that CFTR expression is downregulated in the myocardial tissue of AF patients and shows a negative correlation with senescence markers. CFTR deficiency has been shown to disrupt mitochondrial redox homeostasis and Ca2+/Cl−balance (Jarosz‐Griffiths et al.2024; Kleme et al.2018). Studies indicate that mitochondrial dysfunction leads to insufficient ATP production and excessive ROS generation, which consequently disrupt intracellular Ca2+homeostasis and sarcolemmal excitability in cardiomyocytes, ultimately triggering AF (He et al.2025). Based on this evidence, we propose that downregulation of CFTR may be associated with exacerbated myocardial senescence and mitochondrial oxidative stress in AF patients.
Reduced CFTR expression has been implicated in skeletal muscle aging and mitochondrial oxidative stress (Chen et al.2025). Concurrently, CFTR dysfunction may contribute to alterations in cardiac function (Duus et al.2024). In our study, we observed that CFTR expression was indeed downregulated in the myocardial tissue of D‐gal‐induced aging mice. Notably, CFTR overexpression attenuated pathological damage in the hearts of these aging mice, alongside mitigating cardiomyocyte senescence phenotypes and mitochondrial oxidative stress. Sustained Ca2+influx can lead to intracellular calcium overload, resulting in mitochondrial membrane damage, altered mitochondrial membrane potential, and reduced ATP production. The PMCA is responsible for extruding calcium ions from the cell (Chen et al.2024). Importantly, CFTR‐dependent calmodulin recruitment determines PMCA4 activity (Madácsy et al.2022). Our in vitro experiments demonstrated that inhibition of PMCA partially reversed the protective effects of CFTR overexpression on cardiomyocyte senescence and mitochondrial oxidative stress, leading to the re‐accumulation of Ca2+within the cytoplasm that was otherwise extruded due to CFTR overexpression. These findings strongly suggest that CFTR is highly likely to restore Ca2+homeostasis in senescent cardiomyocytes by activating PMCA, thereby alleviating mitochondrial damage and the senescent phenotype.
Deubiquitination refers to the process by which a series of deubiquitinating enzymes (DUBs) recognize ubiquitin‐tagged proteins and hydrolyze and remove ubiquitin chains from specific amino acid residues on substrate proteins (Zhan et al.2024). Deubiquitination is considered as crucial as its counterpart, ubiquitination, in maintaining the half‐life, activity, and localization of proteins under both normal and pathological conditions (Bhattacharya et al.2020). Deubiquitinating enzymes can stabilize the CFTR protein by removing polyubiquitin chains (Qian et al.2025). In this study, we identified USP45 as the deubiquitinating enzyme for CFTR in cardiomyocytes, which specifically targets K48‐linked ubiquitin chains and modifies the K688 residue of CFTR. Knockdown of USP30 has been shown to alleviate D‐gal‐induced mitochondrial damage and inhibit cardiomyocyte senescence (Pan et al.2021). Similarly, USP11 stabilizes murine double minute 2 (MDM2) in a manner dependent on its deubiquitinase activity, thereby suppressing cellular senescence (Kim et al.2024). These studies indicate that deubiquitinating enzymes are involved in regulating cellular senescence, and their effects depend on their specific protein substrates. Both in vivo and in vitro experiments in this study demonstrate that USP45 can mitigate D‐gal‐induced cardiomyocyte senescence and mitochondrial oxidative stress damage by stabilizing CFTR.
Although this study reveals a novel mechanism through which USP45 stabilizes the CFTR protein by deubiquitinating it at the K688 residue and K48‐linked ubiquitin chains, thereby activating PMCA to restore calcium homeostasis and alleviate mitochondrial oxidative stress and cardiomyocyte senescence, certain limitations remain. The clinical sample size used in this study is relatively small, and further validation in a larger cohort study is needed. The D‐gal‐induced aging model, while classic, cannot fully replicate the multifactorial and complex process of natural human cardiac aging. Furthermore, the precise molecular mechanism of the interaction between CFTR and PMCA, and whether USP45 has additional targets in cardiac aging, requires further elucidation. The current intervention strategy of gene overexpression also remains distant from clinical translation. To address these issues, future research will first validate the generalizability of this pathway in various models, including naturally aged mice. We will employ techniques such as co‐immunoprecipitation and fluorescence resonance energy transfer (FRET) to dissect the formation mechanism of the CFTR‐PMCA complex. Concurrently, we will focus on screening small‐molecule compounds or gene therapy strategies that can specifically activate the USP45‐CFTR pathway, assessing their cardiac specificity and long‐term safety. Finally, by analyzing the correlation between the expression of USP45 and CFTR and cardiac function in human heart tissues across different age groups, we aim to translate these findings towards clinical application, providing a new theoretical foundation and potential therapeutic targets for delaying cardiac aging. Given the potential variability in TEM imaging planes, future research should utilize quantitative software for myofiber orientation analysis to achieve a more objective evaluation of myofiber organization. Furthermore, future studies should incorporate morphometric analysis to systematically quantify cardiomyocyte cross‐sectional area, thereby enabling a more comprehensive assessment of the relationship between CFTR regulation and cardiac structural remodeling.
Conclusion
In summary, this study demonstrates that USP45 enhances CFTR protein stability via deubiquitination, thereby activating PMCA to alleviate D‐gal‐induced mitochondrial oxidative stress and cardiomyocyte senescence. These findings provide a novel therapeutic strategy for intervening in cardiovascular diseases associated with cardiac aging.
Author Contributions
Chun Chen, Longtan Jiang, Yuewen Qiu, Chong Liu:data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review and editing.Chun Chen:data curation, formal analysis.Xiao Long,Pengcui Wu, Liang Li, Haixia Xu:data curation.Li Yang:data curation, resources, writing – review and editing.Ping Deng:resources, supervision, writing – review and editing.Haixia Xu:investigation, resources writing – review and editing.Qiao Jin:conceptualization, data curation, project administration, resources, supervision, writing – review and editing.
Funding
The work was supported by Natural Science Foundation of Hunan Province (Nos. 2025JJ80555, 2026JJ30184, 2025JJ80562, 2026JJ81629) and Natural Science Foundation of Changsha City (No. kq2502323).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Adili, A. , X. Zhu, H. Cao, et al. 2022. “Atrial Fibrillation Underlies Cardiomyocyte Senescence and Contributes to Deleterious Atrial Remodeling During Disease Progression. ”Aging and Disease13, no. 1: 298–312. . doi.org/10.14336/ad.2021.0619
- Akita, H. , S. Yoshie, T. Ishida, Y. Takeishi, andA. Hazama. 2020. “Negative Chronotropic and Inotropic Effects of Lubiprostone on iPS Cell‐Derived Cardiomyocytes via Activation of CFTR. ”BMC Complementary Medicine and Therapies20, no. 1: 118. . doi.org/10.1186/s12906-020-02923-6
- Artusi, I. , M. Rubin, andG. Cozza. 2025. “Redox Imbalance in Cystic Fibrosis: The Multifaceted Role of Oxidative Stress. ”Pharmaceuticals (Basel)18, no. 6: 784. . doi.org/10.3390/ph18060784
- Bhattacharya, U. , F. Neizer‐Ashun, P. Mukherjee, andR. Bhattacharya. 2020. “When the Chains Do Not Break: The Role of USP10 in Physiology and Pathology. ”Cell Death & Disease11, no. 12: 1033. . doi.org/10.1038/s41419-020-03246-7
- Camacho‐Encina, M. , L. K. Booth, R. E. Redgrave, O. Folaranmi, I. Spyridopoulos, andG. D. Richardson. 2024. “Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease. ”Cells13, no. 4: 353. . doi.org/10.3390/cells13040353
- Chen, F. , K. Xu, Y. Han, et al. 2024. “Mitochondrial Dysfunction in Pancreatic Acinar Cells: Mechanisms and Therapeutic Strategies in Acute Pancreatitis. ”Frontiers in Immunology15: 1503087. . doi.org/10.3389/fimmu.2024.1503087
- Chen, Z. , J. Xu, P. Hu, et al. 2025. “Defective Cystic Fibrosis Transmembrane Conductance Regulator Accelerates Skeletal Muscle Aging by Impairing Autophagy/Myogenesis. ”Journal of Cachexia, Sarcopenia and Muscle16, no. 1: e13708. . doi.org/10.1002/jcsm.13708
- Duan, J. , X. Liu, S. Shen, et al. 2023. “Trophoblast Stem‐Cell‐Derived Exosomes Alleviate Cardiotoxicity of Doxorubicin via Improving Mfn2‐Mediated Mitochondrial Fusion. ”Cardiovascular Toxicology23, no. 1: 23–31. . doi.org/10.1007/s12012-022-09774-2
- Duus, L. S. , M. Dons, R. F. Thudium, et al. 2024. “Cardiac Structure and Function in People With Cystic Fibrosis. ”Journal of Cystic Fibrosis23, no. 6: 1138–1145. . doi.org/10.1016/j.jcf.2024.09.012
- Halasi, M. , B. Pandit, andA. L. Gartel. 2014. “Proteasome Inhibitors Suppress the Protein Expression of Mutant p53. ”Cell Cycle13, no. 20: 3202–3206. . doi.org/10.4161/15384101.2014.950132
- Hao, Y. , andW. Liu. 2023. “Metabolic Changes in Cardiac Aging. ”Reviews in Cardiovascular Medicine24, no. 3: 82. . doi.org/10.31083/j.rcm2403082
- He, J. K. , X. X. Jiang, S. Y. Dai, et al. 2025. “β‐Hydroxybutyrate and Citrate Synthase as Potential Diagnostic Biomarkers in Aging‐Related Atrial Fibrillation. ”Journal of Cardiovascular Translational Research18, no. 1: 133–145. . doi.org/10.1007/s12265-024-10569-9
- Hou, Y. , C. Huang, Z. Huang, J. Huang, andB. Zhu. 2024. “STUB1 Exacerbates Calcium Oxalate‐Induced Kidney Injury by Modulating Reactive Oxygen Species‐Mediated Cellular Autophagy via Regulating CFTR Ubiquitination. ”Urolithiasis52, no. 1: 55. . doi.org/10.1007/s00240-024-01547-6
- Jansen, H. J. , M. D. McRae, D. D. Belke, andR. A. Rose. 2025. “Chronic Angiotensin‐Converting Enzyme Inhibition Attenuates Frailty and Protects Against Atrial Fibrillation in Aging Mice. ”Heart Rhythm22, no. 2: 452–465. . doi.org/10.1016/j.hrthm.2024.07.016
- Jarosz‐Griffiths, H. H. , L. R. Caley, S. Lara‐Reyna, et al. 2024. “Heightened Mitochondrial Respiration in CF Cells Is Normalised by Triple CFTR Modulator Therapy Through Mechanisms Involving Calcium. ”Heliyon10, no. 20: e39244. . doi.org/10.1016/j.heliyon.2024.e39244
- Kamada, Y. , H. Tateishi, U. Nakayamada, et al. 2023. “UBE3C Facilitates the ER‐Associated and Peripheral Degradation of Misfolded CFTR. ”Cells12, no. 23: 2741. . doi.org/10.3390/cells12232741
- Kim, W. J. , A. Basit, andJ. H. Lee. 2024. “USP11 Modulates Mitotic Progression and Senescence by Regulating the p53‐p21 Axis Through MDM2 Deubiquitination. ”Biochemical and Biophysical Research Communications726: 150275. . doi.org/10.1016/j.bbrc.2024.150275
- Kleme, M. L. , A. Sané, C. Garofalo, et al. 2018. “CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells. ”Nutrients10, no. 7: 836. . doi.org/10.3390/nu10070836
- Liang, X. , Y. Ding, Y. Zhang, et al. 2015. “Activation of NRG1‐ERBB4 Signaling Potentiates Mesenchymal Stem Cell‐Mediated Myocardial Repairs Following Myocardial Infarction. ”Cell Death and Disease6, no. 5: e1765. . doi.org/10.1038/cddis.2015.91
- Liu, F. , A. L. Kaplan, J. Levring, et al. 2024. “Structure‐Based Discovery of CFTR Potentiators and Inhibitors. ”Cell187, no. 14: 3712–3725. e3734. . doi.org/10.1016/j.cell.2024.04.046
- Liu, Y. , Z. Lin, M. Liu, et al. 2020. “CFTR Deficiency Causes Cardiac Dysplasia During Zebrafish Embryogenesis and Is Associated With Dilated Cardiomyopathy. ”Mechanisms of Development163: 103627. . doi.org/10.1016/j.mod.2020.103627
- Madácsy, T. , Á. Varga, N. Papp, et al. 2022. “Impaired Regulation of PMCA Activity by Defective CFTR Expression Promotes Epithelial Cell Damage in Alcoholic Pancreatitis and Hepatitis. ”Cellular and Molecular Life Sciences79, no. 5: 265. . doi.org/10.1007/s00018-022-04287-1
- Maggiorani, D. , Y. Santin, K. Formoso, et al. 2024. “Identification of Prominin‐2 as a New Player of Cardiomyocyte Senescence in the Aging Heart. ”Aging Cell23, no. 9: e14204. . doi.org/10.1111/acel.14204
- Okiyoneda, T. , C. Borgo, V. Bosello Travain, N. Pedemonte, andM. Salvi. 2024. “Targeting Ubiquitination Machinery in Cystic Fibrosis: Where Do We Stand?”Cellular and Molecular Life Sciences81, no. 1: 271. . doi.org/10.1007/s00018-024-05295-z
- Okiyoneda, T. , G. Veit, R. Sakai, et al. 2018. “Chaperone‐Independent Peripheral Quality Control of CFTR by RFFL E3 Ligase. ”Developmental Cell44, no. 6: 694–708. e697. . doi.org/10.1016/j.devcel.2018.02.001
- Pan, W. , Y. Wang, X. Bai, et al. 2021. “Deubiquitinating Enzyme USP30 Negatively Regulates Mitophagy and Accelerates Myocardial Cell Senescence Through Antagonism of Parkin. ”Cell Death Discovery7, no. 1: 187. . doi.org/10.1038/s41420-021-00546-5
- Qian, C. , Z. Wang, Y. Xiong, et al. 2025. “Harnessing the Deubiquitinase USP1 for Targeted Protein Stabilization. ”Journal of the American Chemical Society147, no. 17: 14564–14573. . doi.org/10.1021/jacs.5c01662
- Ramaccini, D. , V. Montoya‐Uribe, F. J. Aan, et al. 2020. “Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. ”Frontiers in Cell and Developmental Biology8: 624216. . doi.org/10.3389/fcell.2020.624216
- Redgrave, R. E. , E. Dookun, L. K. Booth, et al. 2023. “Senescent Cardiomyocytes Contribute to Cardiac Dysfunction Following Myocardial Infarction. ”NPJ Aging9, no. 1: 15. . doi.org/10.1038/s41514-023-00113-5
- Rubin, M. , I. Artusi, andG. Cozza. 2025. “Mapping the Oxidative Landscape in Cystic Fibrosis: Methodological Frontiers and Application. ”Frontiers in Pharmacology16: 1632924. . doi.org/10.3389/fphar.2025.1632924
- Scialò, F. , G. Cernera, L. Polise, G. Castaldo, F. Amato, andV. R. Villella. 2024. “Effect of CFTR Modulators on Oxidative Stress and Autophagy in Non‐CFTR‐Expressing Cells. ”International Journal of Molecular Sciences25, no. 19: 10360. . doi.org/10.3390/ijms251910360
- Sellers, Z. M. , V. De Arcangelis, Y. Xiang, andP. M. Best. 2010. “Cardiomyocytes With Disrupted CFTR Function Require CaMKII and Ca(2+)‐Activated Cl(−) Channel Activity to Maintain Contraction Rate. ”Journal of Physiology588, no. Pt 13: 2417–2429. . doi.org/10.1113/jphysiol.2010.188334
- Shaw, J. R. , J. M. Bomberger, J. VanderHeide, et al. 2010. “Arsenic Inhibits SGK1 Activation of CFTR Cl‐ Channels in the Gill of Killifish, Fundulus heteroclitus. ”Aquatic Toxicology98, no. 2: 157–164. . doi.org/10.1016/j.aquatox.2010.02.001
- Song, Y. , B. Spurlock, J. Liu, andL. Qian. 2024. “Cardiac Aging in the Multi‐Omics Era: High‐Throughput Sequencing Insights. ”Cells13, no. 20: 1683. . doi.org/10.3390/cells13201683
- Stefanatos, R. , andA. Sanz. 2018. “The Role of Mitochondrial ROS in the Aging Brain. ”FEBS Letters592, no. 5: 743–758. . doi.org/10.1002/1873-3468.12902
- Tang, X. , P. H. Li, andH. Z. Chen. 2020. “Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. ”Frontiers in Endocrinology (Lausanne)11: 280. . doi.org/10.3389/fendo.2020.00280
- Taniguchi, S. , R. Fukuda, andT. Okiyoneda. 2023. “The Multiple Ubiquitination Mechanisms in CFTR Peripheral Quality Control. ”Biochemical Society Transactions51, no. 3: 1297–1306. . doi.org/10.1042/bst20221468
- Tepp, K. , M. Puurand, N. Timohhina, et al. 2017. “Changes in the Mitochondrial Function and in the Efficiency of Energy Transfer Pathways During Cardiomyocyte Aging. ”Molecular and Cellular Biochemistry432, no. 1–2: 141–158. . doi.org/10.1007/s11010-017-3005-1
- Varney, M. E. , M. Niederkorn, H. Konno, et al. 2015. “Loss of Tifab, a Del(5q) MDS Gene, Alters Hematopoiesis Through Derepression of Toll‐Like Receptor‐TRAF6 Signaling. ”Journal of Experimental Medicine212, no. 11: 1967–1985. . doi.org/10.1084/jem.20141898
- Wang, L. , X. Q. Tang, Y. Shi, et al. 2023. “Tetrahydroberberrubine Retards Heart Aging in Mice by Promoting PHB2‐Mediated Mitophagy. ”Acta Pharmacologica Sinica44, no. 2: 332–344. . doi.org/10.1038/s41401-022-00956-w
- Wang, Y. , J. Zhao, Y. Cai, andH. J. Ballard. 2020. “Cystic Fibrosis Transmembrane Conductance Regulator‐Dependent Bicarbonate Entry Controls Rat Cardiomyocyte ATP Release via pannexin1 Through Mitochondrial Signalling and Caspase Activation. ”Acta Physiologica (Oxford, England)230, no. 1: e13495. . doi.org/10.1111/apha.13495
- Wellmerling, J. H. , S. W. Chang, E. Kim, et al. 2019. “Reduced Expression of the Ion Channel CFTR Contributes to Airspace Enlargement as a Consequence of Aging and in Response to Cigarette Smoke in Mice. ”Respiratory Research20, no. 1: 200. . doi.org/10.1186/s12931-019-1170-3
- Wu, F. , R. Hu, X. Huang, et al. 2023. “CFTR Potentiator Ivacaftor Protects Against Noise‐Induced Hair Cell Loss by Increasing Nrf2 and Reducing Oxidative Stress. ”Biomedicine & Pharmacotherapy166: 115399. . doi.org/10.1016/j.biopha.2023.115399
- Zhan, X. , Y. Yang, Q. Li, andF. He. 2024. “The Role of Deubiquitinases in Cardiac Disease. ”Expert Reviews in Molecular Medicine26: e3. . doi.org/10.1017/erm.2024.2
- Zhang, S. , B. Qiu, B. Lv, et al. 2024. “Endogenous Sulfur Dioxide Deficiency as a Driver of Cardiomyocyte Senescence Through Abolishing Sulphenylation of STAT3 at Cysteine 259. ”Redox Biology71: 103124. . doi.org/10.1016/j.redox.2024.103124
- Zhang, Y. P. , Y. Zhang, Z. B. Xiao, et al. 2018. “CFTR Prevents Neuronal Apoptosis Following Cerebral Ischemia Reperfusion via Regulating Mitochondrial Oxidative Stress. ”Journal of Molecular Medicine (Berlin, Germany)96, no. 7: 611–620. . doi.org/10.1007/s00109-018-1649-2
- Zhao, Y. , Z. Wang, D. Feng, et al. 2019. “p66Shc Contributes to Liver Fibrosis Through the Regulation of Mitochondrial Reactive Oxygen Species. ”Theranostics9, no. 5: 1510–1522. . doi.org/10.7150/thno.29620
Republished from the open web under CC-BY. Authors: Chen C, Jiang L, Qiu Y, Liu C, Long X, Wu P, Li L, Xu H, Deng P, Yang L, Jin Q. Read the original.