Elliptical β-barrel deformation underlies gating in VDAC1.
Gating by voltage-dependent anion channels (VDAC) regulates mitochondrial metabolite flux, yet the structural mechanism underlying the open-to-closed transition remains unresolved. Here, we combine atomistic molecular dynamics (MD) simulations with double electron-electron resonance (DEER), using hydrostatic pressure as a reversible thermodynamic perturbation to shift conformational equilibria and stabilize low-population states. MD simulations reveal localized intrinsic flexibility within β-strands β1-β5 and β19, as well as in cytosolic loops connecting β6-β7 and β8-β9. High-pressure DEER measurements in lipid nanodiscs corroborate these predictions, identifying reversible, pressure-dependent distance changes within the pore lumen consistent with asymmetric deformation of the β-barrel. DEER-informed analysis of unbiased MD trajectories reveals an elliptical β-barrel conformation aligned parallel to the N-terminal helix that corresponds to the pressure-stabilized experimental state. ATP permeation simulations identify a free-energy barrier to metabolite translocation in this elliptical geometry, whereas diffusion through the circular open state is energetically favorable. These findings indicate that the elliptical conformation represents a transient gating-competent state rather than a fully closed channel. Together, our results support a gating mechanism driven by reversible β-barrel deformation and establish pressure-perturbed DEER integrated with MD as a general strategy for capturing transient, functionally relevant conformations of membrane channels.
INTRODUCTION
Embedded in the mitochondrial outer membrane (MOM), the voltage‐dependent anion channel (VDAC) serves as the primary conduit for ion and metabolite exchange between the cytosol and mitochondria, facilitating the transport of ATP, ADP, pyruvate, and other metabolites essential for oxidative phosphorylation and cellular bioenergetics (Bergdoll et al.,2017; Rostovtseva & Colombini,1997). VDAC is a dynamic channel, transitioning between a well‐defined single open state and multiple distinct closed states, exerting profound control over mitochondrial activity (Lemasters & Holmuhamedov,2006; Queralt‐Martin et al.,2019). In the open state, VDAC supports efficient metabolite flux across the MOM, whereas closure sharply restricts metabolite transport and limits ATP and ADP exchange (Rostovtseva & Colombini,1996). Through these transitions, VDAC directly regulates mitochondrial energy output. Despite the importance of this process, the structural mechanism underlying VDAC gating remains unresolved. Understanding this mechanism is particularly important given the involvement of VDAC dysfunction in cancers (Heslop et al.,2021; Yuan et al.,2025), neurodegenerative diseases (Verma et al.,2022), and metabolic disorders (Varughese et al.,2021).
High‐resolution structures of VDAC in the open conformation revealed a 19‐strandβ‐barrel architecture containing an N‐terminalα‐helix positioned within the pore lumen (Bayrhuber et al.,2008; Hiller et al.,2008; Ujwal et al.,2008). In contrast, the structure of the closed state has yet to be determined, but early models proposed that voltage gating involves large‐scale displacement or partial unfolding of the N‐terminal segment. However, electrophysiological measurements and crosslinking studies have predominantly disfavored this mechanism, instead implicating structural plasticity of theβ‐barrel itself, including elliptical deformation, as a contributor to gating (Teijido et al.,2012; Zachariae et al.,2012). Consistent with this view, recent molecular‐dynamics simulations indicate localizedβ‐strand flexibility rather than wholesales displacement of the N‐terminal helix (Ngo et al.,2022). While large N‐terminal ejection from the pore is unlikely, smaller rearrangements, such as helix rotation or subtle barrel deformation, remain possible and may occur either independently or in concert. Discriminating between N‐terminal rearrangement andβ‐barrel deformation therefore remains a central challenge, particularly because the closed state is transient, sparsely populated, and typically accessed only under perturbative conditions such as applied voltage, reduced pH, mechanical stress, or interaction with partner proteins (Colombini,2004; Rostovtseva et al.,2006; Teijido et al.,2014).
Here, we combine molecular‐dynamics simulations with double electron–electron resonance (DEER) to probe conformational changes associated with VDAC1 gating. DEER measures nanometer‐scale distances between site‐directed spin labels and is well suited for resolving conformational ensembles of membrane proteins in lipidic or micellar environments. To stabilize low‐population states relevant to gating, we applied hydrostatic pressure, a perturbation known to shift structural equilibria toward conformations with reduced partial molar volume (Akasaka,2003; Lerch et al.,2014,2020). This approach allows direct experimental access to otherwise elusive VDAC conformations and enables quantitative evaluation. Comparison with MD simulations provides atomistic insight into rare conformations of VDAC which may contribute to gating. Using this combined strategy, we find that VDAC1 retains a globally stableβ‐barrel architecture, in the absence of destabilizing mutations, while undergoing reversible, localized conformational changes that produce ellipsoidal barrel geometries. These deformations restrict the pore and are consistent with functional closed‐state behavior (i.e., reduced ion conductance and obstructed ATP flux), providing experimental support forβ‐barrel deformation as a central element of VDAC gating.
RESULTS
MDsimulations identify localized flexibility in the openVDAC1β‐barrel
To characterize the conformational landscape accessible to VDAC1 in its open state, we performed all‐atom molecular dynamics (MD) simulations to identify regions exhibiting elevated intrinsic flexibility. VDAC1 adopts a 19‐strandedβ‐barrel architecture with an N‐terminalα‐helical segment positioned within the pore and oriented parallel to the membrane surface within the pore lumen (Bayrhuber et al.,2008; Hiller et al.,2008; Ujwal et al.,2008). Notably, VDAC1 is the first membrane‐integratedβ‐barrel protein identified with an odd number of strands, an architecture proposed to confer enhanced conformational plasticity and support gating transitions (Teijido et al.,2012; Zeth & Thein,2010). We therefore simulated VDAC1 embedded in a lipid bilayer representative of the mitochondrial outer membrane, with the goal of sampling conformational heterogeneity and identifying structural elements most likely to participate in gating‐associated rearrangements.
Figure1displays backbone root‐mean‐square fluctuations (RMSF) calculated over a 500‐ns simulation, which report the average positional deviation of each residue and provide a measure of local flexibility. While the N‐terminal helix andβ‐strandsβ6–β18 remained comparatively rigid, elevated RMSF values were observed on one side of the barrel, encompassingβ‐strandsβ1–β5 and the structurally contiguousβ19 strand. In addition, pronounced fluctuations were detected in cytosolic loops connectingβ‐strands 6–7 [L(6–7)cyt] andβ‐strands 8–9 [L(8–9)cyt]. Notably, these cytosolic loops displayed greater flexibility than loops facing the intermembrane side of the channel, consistent with their proposed role in accommodating interactions with cytosolic partner proteins (Rosencrans et al.,2025; Rostovtseva & Bezrukov,2008). These features were reproducible across six independent simulations (Figure1; SFig.1), indicating that enhanced flexibility in these regions reflects intrinsic properties of the VDAC1 structure rather than stochastic fluctuations.
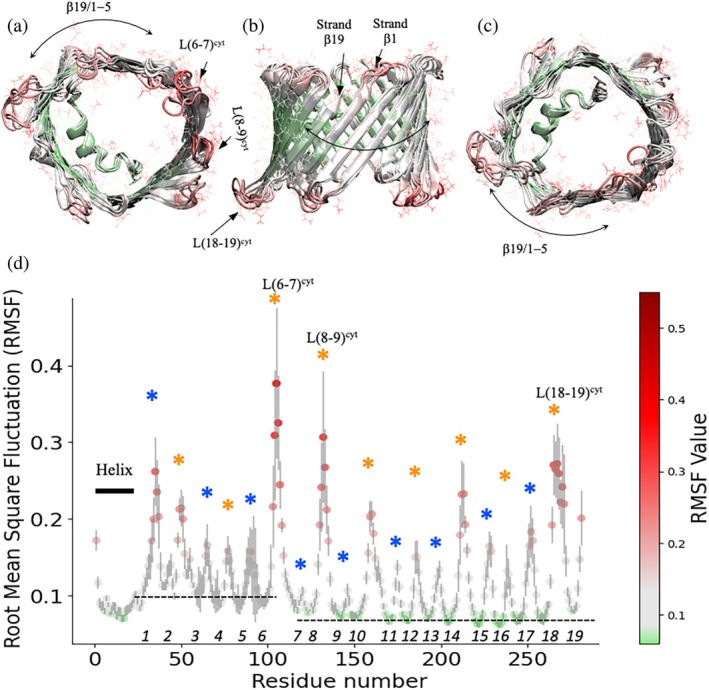
Localized flexibility of VDAC1 revealed by all‐atom molecular dynamics simulations. Five representative structures of VDAC1 sampled over a 500‐ns all‐atom molecular dynamics simulation are shown and color‐coded according to backbone root‐mean‐square fluctuations (RMSF). Structures are viewed from (a) the cytoplasmic side, (b) theβ19–β1–β5 face, and (c) the intermembrane space (IMS) side. Elevated RMSF values are localized to one side of theβ‐barrel, encompassingβ‐strandsβ1–β5 and the structurally contiguousβ19 strand, as well as select cytosolic loop regions. (d) RMSF values for all residues (backbone atoms only) plotted as a function of residue number. Cytosolic loops are indicated by orange asterisks, and IMS‐facing loops by blue asterisks. Numbers on top of thex‐axis represent theβ‐strands. Regions corresponding to the N‐terminal helix exhibit comparatively low RMSF values, consistent with a globally stableβ‐barrel scaffold and localized conformational flexibility.
This pattern of flexibility closely mirrors experimental observations from crystallographic studies. A recent comparison of B‐factors from cryogenic and room‐temperature crystal structures of VDAC1 revealed the largest temperature‐dependent differences in the intracellular portions ofβ‐strandsβ1,β2,β5, andβ6, as well as in cytosolic loop regions L(6–7)cytand L(8–9)cyt(Gonzalez‐DeWhitt et al.,2024). The concordance between MD‐derived RMSF profiles and experimentally observed B‐factor variations supports a consistent picture in which VDAC1 maintains a globally stableβ‐barrel scaffold while exhibiting localized flexibility in specificβ‐strands and cytosolic loops. These regions therefore represent prime candidates for conformational rearrangements associated with VDAC gating and provide a structural framework for the selection and interpretation of site‐directed spin labeling (SDSL) positions used in subsequent DEER experiments.
Structural analysis in lipidic and micellar environments
To quantify conformational flexibility across defined regions of the VDAC1 pore, we employed SDSL and DEER spectroscopy. Six double‐cysteine variants were generated to probe distinct structural regions: the cytosolic surfaceβ2cyt–β11cytandβ6cyt–β15cyt, the mid‐pore regionα2–β13midandβ1mid–β9mid, and the intermembrane space (ims) side withβ4ims–β14imsandβ7ims–β17ims(Figure2). Expected DEER distances were calculated from the murine VDAC1 structure (PDB3EMN) using MD with dummy spin labels (MDDS) (Qi et al.,2020), a computational approach that incorporates spin‐labels in protein structural models and derives distance distributions from short MD simulations. These predictions provide reference values for interpreting experimental DEER data.
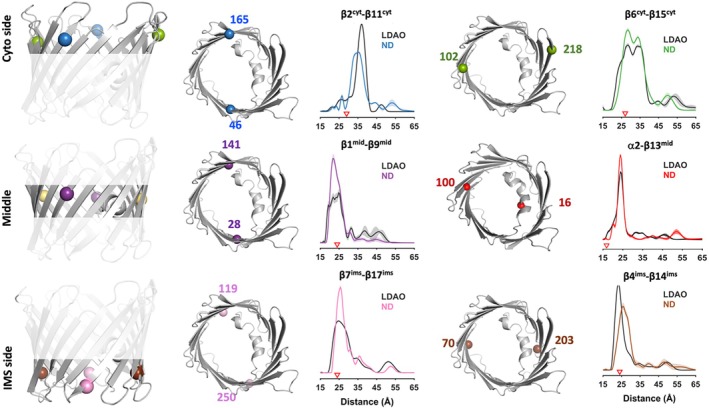
Comparison of VDAC1 structure in lipid nanodiscs and detergent micelles by DEER spectroscopy. SDSL and DEER spectroscopy were used to compare conformational properties of VDAC1 reconstituted in lipid nanodiscs (ND) or solubilized in LDAO micelles. Six double‐cysteine variants were introduced at strategic locations spanning the cytosolic faceβ2cyt–β11cyt,β6cyt–β15cyt, the mid‐poreα2–β13mid,β1mid–β9mid, and the intermembrane space (ims) sideβ4ims–β14ims,β7ims–β17ims. For each pair, the modeled spin‐label positions are shown on the VDAC1 structure (PDB:3EMN), alongside the corresponding DEER‐derived distance distributions for LDAO (black) and nanodiscs (colored traces). Red triangles indicate MDDS‐predicted distances derived from the crystal structure. Associated dipolar evolution data are shown in SFig.2.
To assess how the membrane environment influences VDAC1 structure and dynamics, we compared DEER measurements from proteins reconstituted into lipid nanodiscs with those obtained from samples solubilized in LDAO micelles. While LDAO is widely used for structural studies of VDAC, nanodiscs provide a native‐like bilayer environment and minimize oligomerization artifacts that can complicate DEER analysis. Across all six spin‐label pairs, the dominant DEER peaks closely matched MDDS‐predicted distances in both detergent and lipidic environments (Figure2and SFig.2), indicating that the crystal structure and both experimental conditions represent the open‐stateβ‐barrel.
Despite this global similarity, nanodisc‐reconstituted samples generally exhibited narrower distance distributions than their detergent‐solubilized counterparts, consistent with reduced conformational dynamics. This effect was particularly evident forβ2cyt–β11cytandβ7ims–β17imsspin pairs (Figure2and SFig.2), suggesting that lipid bilayer interactions stabilize specific regions of the barrel. In contrast, the pairβ4ims–β14imsdisplayed modest broadening in nanodiscs, indicative of enhanced flexibility at the IMS interface in a bilayer environment.
Together, these results demonstrate that while detergent and nanodisc environments yield highly similar average structures, lipid bilayers modestly dampen conformational heterogeneity and provide a more stable platform for probing subtle, pressure‐induced rearrangements of VDAC1.
Pressure perturbation reveals conformational changes inVDAC1
Under hydrostatic pressure, LDAO‐solubilized samples exhibited reversible oligomerization that interfered with interpretation of single‐channel behavior (SFig.3). This oligomerization introduces intermolecular DEER contributions that obscure intramolecular distance changes and preclude reliable analysis of conformational states. For this reason, all subsequent pressure‐perturbation experiments were performed exclusively using nanodisc‐reconstituted VDAC1. To probe alternate conformations potentially involved in VDAC1 gating, we subjected nanodisc‐reconstituted samples to 3 kbar hydrostatic pressure. Application of pressure favors protein conformations with reduced partial molar volume by promoting side‐chain repacking and changes in hydration, thereby shifting the structural equilibrium toward low‐population states. Hydrostatic pressure has proven particularly effective for revealing conformational heterogeneity and sparsely populated intermediates in membrane proteins, including pressure‐induced shifts between functional states (Gonzalez‐DeWhitt et al.,2024; Lerch et al.,2020) that are not detectable under ambient conditions, and is used here as a non‐physiological perturbation to probe the conformational landscape rather than to represent a pressure‐driven biological mechanism. Consistent with this, the DEER distance distributions reflect pressure‐induced population shifts within a heterogeneous conformational ensemble, observed as broadening and asymmetric redistribution rather than a uniform shift in average distances.
Application of 3 kbar hydrostatic pressure revealed significant distance changes at four of the six positions (Figure3):β6cyt–β15cyt,β1mid–β9mid,α2–β13mid, andβ4ims–β14ims. The remaining spin pairs—β2cyt–β11cytandβ7ims–β17ims—were largely unchanged, consistent with a rigidβ‐barrel scaffold in these regions. Pressure broadened the distance distributions forβ6cyt–β15cytandβ4ims–β14ims, indicating increased flexibility at both membrane interfaces. Notably, the two mid‐pore pairs shifted in opposite directions, withβ1mid–β9midshifting toward longer distances andα2–β13midtoward shorter distances. These asymmetric changes are consistent with a pressure‐stabilized deformation of the barrel cross‐section at the mid‐pore. All distance changes were reversible upon pressure release (SFig.4), confirming that these conformations coexist with the open state under ambient conditions.
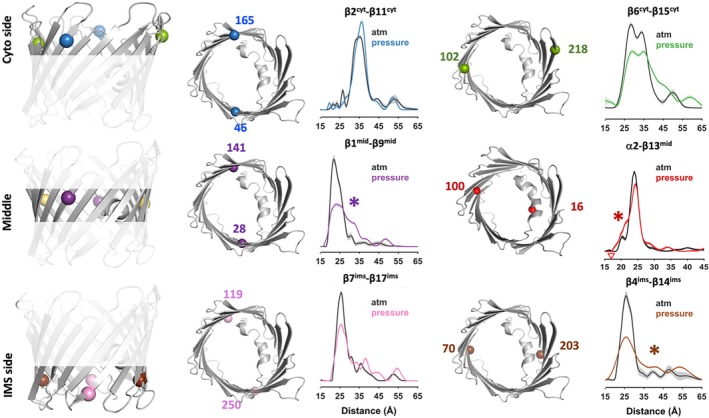
Pressure perturbation reveals local conformational changes in VDAC1. SDSL and DEER spectroscopy were used to monitor pressure‐induced conformational changes in nanodisc‐reconstituted VDAC1 at 3 kbar hydrostatic pressure. Six double‐cysteine mutant pairs were introduced at strategic locations spanning the cytosolic faceβ2cyt–β11cyt,β6cyt–β15cyt, the mid‐poreα2mid–β13mid,β1mid–β9mid, and the intermembrane space (IMS) sideβ4ims‐14ims,β7ims–17ims. For each spin pair, DEER‐derived distance distributions are shown at atmospheric pressure (black) and under 3 kbar pressure (colored traces). Asterisks () indicate spin pairs exhibiting significant but reversible pressure‐induced changes. Associated dipolar evolution data are shown in SFig.9.*
The observation that pressure selectively stabilizes asymmetric mid‐pore rearrangements, while leaving other regions unchanged, supports a model in which VDAC gating involves structural adaptation rather than global unfolding. Such pressure‐stabilized asymmetry is consistent withβ‐barrel deformation mechanisms previously proposed on the basis of electrophysiological measurements (Teijido et al.,2012) and MD simulations (Zachariae et al.,2012).
The N‐terminal domain of VDAC1, comprising theα1 andα2 segments, resides within the pore and has long been implicated in channel gating through its influence on pore electrostatics and barrel stability (Mertins et al.,2012; Teijido et al.,2012). Recent MD simulations by Ngo et al. demonstrated that charged residues within theα2 segment participate in a voltage‐sensitive electrostatic network, and that perturbation of this network induces subtle deformations of theβ‐barrel sufficient to promote channel closure (Ngo et al.,2022). Importantly, this mechanism does not require extensive movement of theα2 segment itself, but instead highlights its role in coupling local electrostatic perturbations to collective barrel rearrangements. Consistent with this model, Najbauer et al. reported destabilization of theα2 region accompanied by smallβ‐barrel deformations during gating in a quintuple VDAC mutant (Najbauer et al.,2022). Together, these studies suggest that theα2 segment, anchored to the barrel wall through hydrophobic interactions, plays a central role in gating by transducing small local perturbations into subtle, collective deformations of theβ‐barrel.
In line with this framework, we measured the wall‐to‐helix distance between a stable mid‐pore barrel position and theα2 segmentα2–β13mid. Under hydrostatic pressure, this distance distribution developed a reproducible ~4 Å shorter shoulder (Figure3) indicating a pressure‐stabilized rearrangement involving theα2 region. While this change could reflect modest helix repositioning or rotation, its magnitude and reversibility are also consistent with local barrel deformation coupled toα2 motion. Because changes in DEER distance distributions may reflect both backbone rearrangements and altered spin‐label rotamers, we used MD simulations to gain structural insight into the conformational changes most consistent with the observed pressure‐induced distance shifts.
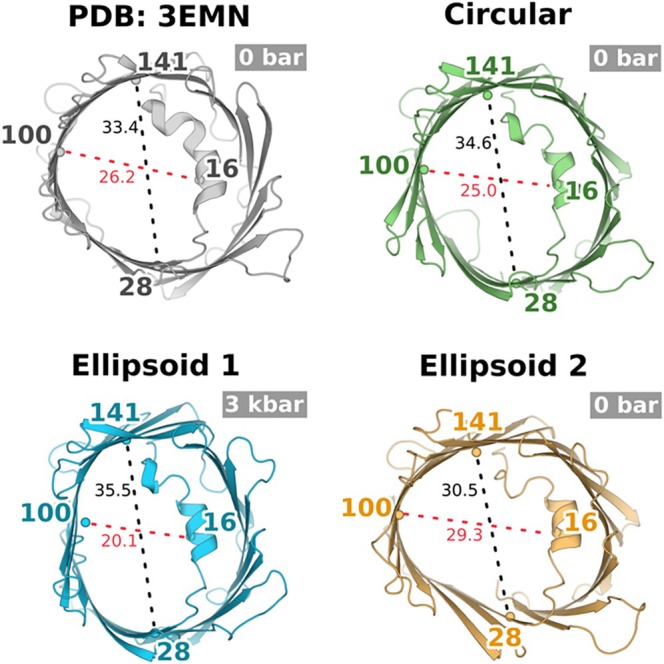
DEER‐consistent MD ensembles reveal elliptical VDAC1 conformations. Average structures representing the near‐circularβ‐barrel conformation (green) and two elliptical conformations—ellipsoid 1 (blue) and ellipsoid 2 (orange) identified by DEER‐informed filtering of unbiased MD simulations—are shown in comparison with the open‐state crystal structure of VDAC1 (gray; PDB3EMN). Distances corresponding to experimentally probed mid‐pore DEER spin‐label pairs are indicated, illustrating axis‐specific barrel deformations. The ellipsoid 1 conformation corresponds to the pressure‐stabilized DEER state, whereas the ellipsoid 2 conformation aligns with the ambient‐pressure DEER state.
DEER‐informed ensemble analysis reveals ellipticalVDAC1conformations
To obtain structural insight into the pressure‐dependent changes observed in the DEER experiments, we applied a DEER‐informed analysis to our MD simulations (Belyaeva & Elgeti,2024). The MD trajectories were run without any applied pressure, voltage, or DEER‐derived restraints and therefore sample the conformational heterogeneity intrinsic to the open‐state energy landscape. Experimental DEER distance distributions were used post hoc to identify subsets of simulation frames most consistent with the experimentally observed pressure‐stabilized or ambient‐pressure states. The DEER‐informed filtering and clustering procedure is summarized in SFig.5.
For the DEER‐informed analysis of the MD trajectories, we used mid‐pore distance constraints fromβ1mid–β9midandα2–β13midvariants that exhibited reproducible, pressure‐dependent shifts in the experimental distributions. For each MD trajectory, spin labels were attached in silico to the simulated frames, and spin–spin DEER distance distributions were simulated using the chiLife Python package (Tessmer & Stoll,2023). The simulated DEER distributions from individual frames were then averaged across each complete trajectory to characterize the overall VDAC1 conformation sampled in that trajectory.
Across five independent MD trajectories, three reproduced the experimental trends observed under either pressure or ambient conditions. One trajectory (T2) yielded distance distributions consistent with the pressure‐stabilized DEER state, whereas two trajectories matched the ambient‐pressure DEER state (T1 and T4). The remaining trajectories did not reproduce distance distributions consistent with the experimental DEER data, and were therefore not used to describe pressure‐associated conformational ensembles. This outcome was expected, as the simulations were not biased in any way to reach low‐population, or pressure‐stabilized conformations.
Because DEER distance distributions reflect contributions from both backbone rearrangements and spin‐label rotameric flexibility, we applied a filtering procedure to minimize the influence of local rotamer effects and emphasize global conformational differences. Simulation frames exhibiting DEER‐consistent distances, characteristic of the pressure‐stabilized population, were selected and subsequently clustered (SFig.5). This analysis ensured that the resulting ensembles reflect coordinated rearrangements of protein segments rather than fluctuations of individual spin labels.
Clustering of the filtered frames revealed three dominant conformational ensembles. The most populated ensemble corresponds to a near‐circularβ‐barrel geometry closely resembling the crystallographic open state (PDB3EMN). Two additional ensembles exhibited elliptical barrel geometries, which we term ellipsoid 1 and ellipsoid 2, with elongation axes parallel or orthogonal to the N‐terminal helix, respectively (Figure4). The ellipsoid 1 ensemble aligned with the pressure‐stabilized DEER distance distributions, whereas the ellipsoid 2 ensemble corresponded to the ambient‐pressure DEER state, consistent with reversible, axis‐specific barrel deformation.
To assess the functional implications of these conformations, we analyzed the pore bottleneck size, profile of the pore radius, and estimated channel conductance using the HOLE2 program (Smart et al.,1996). The ellipsoid 1 ensemble exhibited a reduced pore bottleneck radius relative to both the circular and ellipsoid 2 ensembles, with a pronounced constriction near the mid‐pore region (Figure5b/d). However, estimated single‐channel conductance values derived from pore geometry differed only modestly between the different ensembles (Figure5c), remaining substantially higher than conductance values typically associated with the “closed‐state” recorded by electrophysiological techniques (Choudhary et al.,2010,2014). Because conductance estimates based on static structural models are sensitive to sampling and may not capture coupled rearrangements beyond the DEER‐informed regions, we interpret these values cautiously. We repeated this analysis using an extended set of frames collected from all five trajectories filtered using only distance criteria (SFig.8). While we observe the same trend, the effect is blurred due to the larger contribution from spin‐label dynamics.
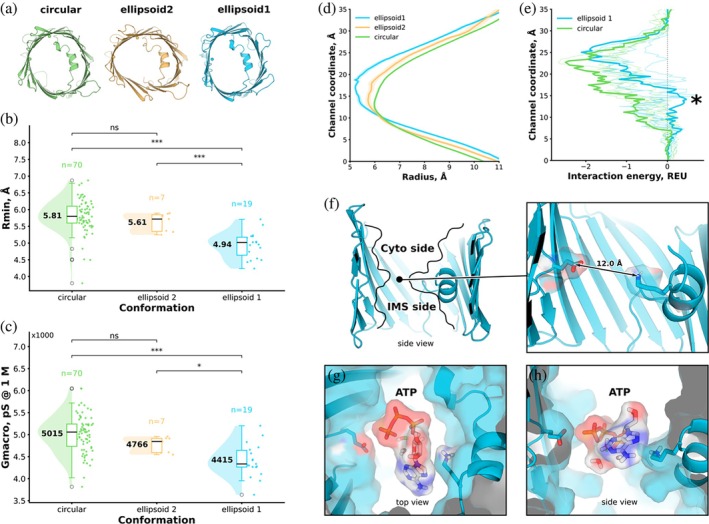
Geometric and functional characteristics of the VDAC1 conformations. (a) Representative structures of the three dominant conformational ensembles identified by DEER‐informed MD analysis: circular (green), ellipsoid 1 (blue), and ellipsoid 2 (orange). (b) Distribution of pore bottleneck radii (R_min) calculated using HOLE2 for each ensemble. Individual data points are representative frames; box plots indicate median and interquartile range, and half‐violin plots show the underlying distributions. The ellipsoid 1 ensemble exhibits a reduced bottleneck radius relative to the circular and ellipsoid 2 ensembles. (c) Estimated single‐channel conductance at 1 M salt concentration derived from pore geometry showing only modest but significant differences for the two ellipsoid conformations. (d) Axial pore‐radius profiles averaged over each ensemble, highlighting a pronounced mid‐pore constriction in the ellipsoid 1 conformation. (e) Predicted ATP interaction energy profiles along the channel axis, calculated using Rosetta LigandPathFinder. The ellipsoid 1 ensemble exhibits a positive free‐energy barrier in the bottleneck region (), whereas the circular ensemble displays a continuous energetically favorable pathway. (f) Side view of the ellipsoid 1 conformations illustrating narrowing of the pore. (g), (h) Representative ATP poses within the ellipsoid 1 channel shown in top (g) and side (h) views, illustrating the bottleneck area.*
In contrast, ATP permeation simulations provided a more direct functional discriminator. Using Rosetta LigandPathFinder, ATP translocation through the ellipsoid 1 ensemble encountered a positive free‐energy barrier at the bottleneck, whereas the circular ensemble displayed a continuous energy funnel consistent with unhindered diffusion (Figure5e). These results identify the ellipsoid 1 conformation as gating‐competent, capable of restricting metabolite permeation, even though it does not fully recapitulate a low‐conductance closed state observed electrophysiologically.
DISCUSSION
This study provides new insight into the structural dynamics of the VDAC, a key MOM protein essential for cellular homeostasis. Althoughβ‐barrel membrane proteins are often regarded as structurally rigid, our combined experimental and computational data reveal that VDAC1 exhibits intrinsic conformational flexibility, consistent with earlier observations from simulation and spectroscopy studies (Najbauer et al.,2022; Choudhary et al.,2010; Ngo et al.,2022; Teijido et al.,2012; Zachariae et al.,2012). VDAC's unusual 19‐strandedβ‐barrel topology may provide a unique balance between global stability and localized flexibility, enabling regulated gating transitions while preserving overall structural integrity.
By integrating atomistic MD simulations with DEER analysis under hydrostatic pressure, we identify reversible elliptical deformation of theβ‐barrel as a defining structural feature associated with channel closure. Mid‐pore reporters (α2–β13mid,β1mid–β9mid) exhibited robust, pressure‐dependent distance shifts that are consistent with asymmetric expansion and compaction along an axis parallel to the N‐terminal segment. These observations indicate that VDAC gating involves coordinated barrel rearrangements rather than global unfolding.
In contrast to earlier models proposing large‐scale displacement or extrusion of the N‐terminal helix into the cytosol (Casadio et al.,2002; Colombini,1989), we observed no evidence for such movements, even at 3 kbar hydrostatic pressure. Helix extrusion would be expected to produce pronounced increase in hydration and distance distributions, which were not detected. Recent work by Daniilidis et al. demonstrates N‐terminal displacement under conditions that promote oligomerization or strong confinement, including the use of small nanodiscs that mimic an oligomeric state (Daniilidis et al.,2025). In contrast, under more native‐like conditions—specifically monomeric VDAC reconstituted in larger nanodiscs—the N‐terminal helix remains positioned within the pore. Our results, obtained in monomeric nanodiscs, do not support large‐scale helix expulsion under these conditions. Moreover, our findings are consistent with prior crosslinking studies demonstrating that tethering the N‐terminus to the barrel wall—such as cysteine crosslinks between V17–A205 or L10–A170—does not abolish channel gating. Together, these results argue against models in which gating is driven primarily by wholesale N‐terminal displacement. Instead, our data support a mechanism centered onβ‐strand rearrangements involvingβ1 and its structural continuation intoβ19 andβ2–β5, producing an elliptical constriction that restricts metabolite translocation while maintaining intrapore positioning of the N‐terminal helix.
We further considered whether barrel rearrangements might arise from disruption of salt bridges, as prior work showed that acidification of the cytosolic side of VDAC1 alters gating behavior, likely by weakening electrostatic interactions. To test this possibility, we collected DEER measurements at pH 3.6 under atmospheric pressure. No significant changes in helix positioning were observed (SFig.6), indicating that hydrostatic pressure–induced conformational changes cannot be mimicked by simple acidification. These findings suggest that the observed pressure‐driven barrel deformation arises primarily from side‐chain repacking, rather than from increased hydration or breakage of salt bridges.
In summary, we propose that VDAC gating occurs through a reversible elliptical deformation of theβ‐barrel rather than by N‐terminal ejection. This mechanism accommodates regulation by diverse physical stimuli—including voltage, pressure, and membrane environment—while preserving the energetic efficiency of a compactβ‐barrel fold. More broadly, our findings highlightβ‐barrel plasticity as an under‐recognized regulatory principle in mitochondrial channels. By responding dynamically to metabolic cues and stress‐related perturbations, VDAC functions as a responsive mitochondrial gatekeeper, integrating bioenergetic demand with mitochondrial function.
METHODS
Molecular dynamics simulations
All‐atom MD simulations were initiated from configurations extracted from previously published Martini coarse‐grained (CG) simulations (Lafargue et al.,2025). These consisted of three independent replicas of membrane systems with identical composition, comprising VDAC1 in a 2:1 POPC:POPE mixture. Two snapshots per system (at 10 and 20 μs) were selected to increase sampling, yielding six replicas derived from independently built systems. This approach captures both stochastic variability (initial velocities) and differences in initial coordinates, including membrane packing and local protein–lipid environments, providing a stringent assessment of reproducibility. From these CG systems, the lipid configurations were retained and transformed into an all‐atom system using the backward procedure (Wassenaar et al.,2014). To maintain high‐resolution quality for the protein, the corresponding “backwarded structure” was replaced by a fresh all‐atom model previously derived from the experimental mouse hVDAC1 structure available at 2.3 Å resolution (PDB ID:3EMN) (Ujwal et al.,2008).
The systems were resized around VDAC to 9.7 × 9.7 nm in thexyplane, with corresponding new periodic boundary conditions. The systems were relaxed to an equilibrium state using a series of energy minimization and constrained MD procedures described in the backward paper. The simulations were run using GROMACS software (Abraham et al.,2015) with the CHARMM36m force field with CMAP corrections (Huang et al.,2017).
The thermalization and equilibration process was developed over a six‐stage procedure where distinct position restraints were sequentially released. The first equilibration run applied restraints of 4000, 2000, and 1000 kJ/mol/nm2to the protein backbone, side chains, and lipid head groups, respectively. In the final equilibration run, only small restraints (50 kJ/mol/nm2) remained on the atoms of the protein backbone. The system was heated to 303.15 K during the first run and controlled by a Berendsen thermostat. Pressure control was set using a Berendsen barostat at 1.0 bar in the second stage, employing semi‐isotropic control (τp= 5 ps and compressibility of 4.5 × 10−5in the membrane plane and its normal direction). The first three equilibration processes ran over 125 ps, while 500 ps were used for the last three. The time step was increased from 1 to 2 fs in the fourth stage, and bond lengths involving hydrogen atoms were maintained constant using the LINCS algorithm throughout the simulations.
For the production period, which lasted over 500 ns, the Berendsen thermal and pressure control algorithms were replaced by the Nosé–Hoover thermostat and Parrinello–Rahman barostat schemes respectively. The remaining position restraints on the protein backbone were removed. The simulations were run on the supercomputer facilities provided by the GENCI at the TGCC (CEA)—Saclay France using GROMACS v2018.
Protein purification
mVDAC1 was produced inEscherichia coliand refolded from inclusion bodies. A detailed protocol is available in Dearden et al. (2024). Briefly, M15E. colicells were used to express mVDAC1 in inclusion bodies. The protein was isolated using a two‐step purification process: solubilized inclusion bodies were purified with a Talon metal affinity column, and the eluted protein was refolded using LDAO. The refolded protein was centrifuged at 200,000 ×gfor 45 min, and DTT was removed using a PD10 desalting column, with buffer exchanged to 20 mM Hepes (pH 7.0), 150 mM NaCl, and 0.1% LDAO to facilitate labeling. The protein was then incubated with a 1:1 molar ratio of the spin label reagent 1‐oxyl‐2,2,5,5‐tetramethylpyrroline‐3‐methyl methanethiosulfonate (MTSSL) for 30 min at 4°C to generate the R1 side chain. The protein was concentrated using an Amicon Ultra‐50k (Millipore) and subjected to size exclusion chromatography (Superdex 200; GE Healthcare) in 150 mM NaCl, 20 mM Tris·HCl (pH 8.0), and 0.1% LDAO to remove excess spin label and misfolded proteins. 20% glycerol‐D8 was added to the sample. All samples for the same mutants were concentration‐normalized (80–100 μM, depending on the mutant) to compare modulation depths and distance distributions.
Reconstitution into nanodiscs
Labeled VDAC was mixed with lipid mix 25 mM Egg PC (avanti lipids) in 50 mM sodium cholate, and membrane scaffold protein without a His‐tag (MSP1D1(−)). We used an 8 times excess of ND/VDAC to make sure we have monomeric VDAC per nanodiscs. The molar ratio of VDAC1/MSP1D1(−)/lipid was 1:16:800 in buffer 20 mM Tris pH 8.5 (to avoid dimerization; Bergdoll et al.,2018), 150 mM NaCl. The mixture was incubated for 60 min at 4°C, and nanodisc assembly was initiated by adding 60% (w/v) Bio‐Beads SM‐2 (Bio‐Rad), followed by incubation for 4–16 h at 4°C with rotation. Assembled nanodiscs containing His‐tagged VDAC protein were isolated by binding to Ni‐NTA resin equilibrated in buffer 20 mM Tris pH 8, 150 mM NaCl and washed with 10 column volumes of buffer to remove empty nanodiscs. Nanodiscs containing VDAC were eluted with 20 mM Tris pH 8, 150 mM NaCl, 300 mM imidazole, concentrated using an Amicon Ultra‐50k (Millipore) and subjected to size exclusion chromatography (column Superdex 200, 10/300; GE Healthcare) in buffer 20 mM Tris‐Cl, pH 8, 150 mM NaCl. 20% glycerol‐D8 was added to the sample. All samples for the same mutants were concentration‐normalized (80–100 μM, depending on the mutant) to compare modulation depths and distance distributions.
DEERexperiment
4‐Pulse DEER experiments were conducted at 50 K on a Q‐band upgraded ElexSys 580 (Bruker, Germany) equipped with an arbitrary waveform generator, a 150 W traveling wave tube amplifier, and a QT‐2 probehead (Bruker, Germany) using the standard dead time free sequence (Pannier et al.,2000). Pulse lengths were optimized using echo nutation experiments and set to 16–20 ns and 32–40 ns for pi/2 and pi‐pulses, respectively. A 100 ns 50 MHz‐chirp pulse was applied as a pump pulse 70 MHz above the observer frequency.
Dipolar evolutions were recorded with 16 ns time resolution and analyzed model‐free in LongDistances v.1112 (Altenbach,2021). The smoothness parameter was determined by L‐curve criterion. Standard deviations for distances were determined using the error analysis implementation in LongDistances using standard parameters and the inherited model‐free parameters.
Simulation ofDEERdistance distributions
Five MD trajectories (500 ns each, 1000 frames per trajectory) were used as input for the analysis. DEER distance distributions were simulated for residue pairs 16/100 and 28/141, selected because experimental measurements at 0 bar and 3 kbar show clear differences in the channel interior (Figure3). At 3 kbar, the 16/100 distribution develops a shoulder in shorter distances area absent under normal conditions, whereas the 28/141 distribution exhibits a broadened tail toward longer distances.
For each frame of every trajectory, DEER distance distributions were simulated using the chiLife (v1.1.6) and MDAnalysis (v2.10.0) Python libraries (Michaud‐Agrawal et al.,2011; Tessmer & Stoll,2023). MDAnalysis was used to iterate through the MD trajectories, while chiLife computed the DEER distribution for each frame. Ensembles of covalently attached spin labels were modeled using the rotamer library approach implemented in chiLife, employing the R1 label used in the original experiments (Tessmer & Stoll,2023). Each per‐frame distance distribution was normalized to unity, as were the reference experimental DEER distributions.
In addition, DEER distance distributions were averaged over all frames for each trajectory, and the standard error of the mean (SEM) was calculated (SFig.7). These averaged simulated distributions were compared with the experimental data to assess which trajectories reproduce the pressure‐dependent trends, namely the emergence of shorter distances for the 16/100 pair and longer distances for the 28/141 pair. Based on the comparison of distributions, trajectories T1, T2, and T4 were used for subsequent analysis. All the code is available on Zenodo repository.
Extraction of peak of simulatedDEERdistribution
To extract a single characteristic peak from each simulated DEER distribution, each per‐frame distribution was first smoothed with a one‐dimensional Gaussian filter (σ = 2; scipy.ndimage.gaussian_filter1d, SciPy v1.16.3) (Virtanen et al.,2020). This step reduces small fluctuations and noise that can create artificial minor peaks. Peaks were then identified using scipy.signal.find_peaks, and the peak with the highest intensity in the smoothed distribution was selected as the main peak for that frame.
DEER‐based filtering ofMDsimulation frames
To identify VDAC1 structures with a barrel shape deviating from the predominantly circular conformation observed under normal conditions (PDB:3EMN), we applied a DEER‐based filtering procedure. Frames showing extreme positions of the main peak in the simulated DEER distributions were selected, capturing contributions from both protein backbone and spin label dynamics.
Two criteria were defined using the previously identified main peak of the per‐frame simulated DEER distributions:d(16/100)≤ 22 Å and d(28/141)≥18 Åd(16/100) ≥26 Å and d(28/141)≤ 17 Å
Frames satisfying criterion 1 were assigned to cluster 1, those satisfying criterion 2 to cluster 2, and frames meeting neither criterion were left unassigned. The selected frame numbers were written to GROMACS index (.ndx) files and used to extract the corresponding structures from trajectories T1, T2, and T4 (Abraham et al.,2015). This yielded six filtered trajectory sets in total. Cluster 1 contains frames in which the VDAC barrel is compressed along the 16/100 direction; cluster 2 includes near‐circular or 28/141‐compressed structures. Trajectories T1 and T4 contributed a larger fraction of frames to cluster 2, while T2 showed the opposite trend.
CAdistance‐based filtering ofMDsimulation frames
The R1 spin label used in the DEER experiments is highly dynamic, making it difficult to separate the dynamics of protein backbone and spin labels (Polyhach et al.,2011; Sezer et al.,2008) in the computational analysis. To isolate the contribution of the dynamics of protein backbone, we applied an additional filtering step based solely on Cα/Cαdistances.
For each frame of trajectories T1, T2, and T4, the Euclidean distance between Cαatoms of residue pairs 16/100 and 28/141 was computed directly from trajectory coordinates using MDAnalysis. In T1, data points cluster near the center of the plot, consistent with a predominantly circular barrel geometry matching the x‐ray structure (PDB:3EMN). T2 shows a shift toward shorter 16/100 and longer 28/141 distances, indicating compression along the 16/100 direction. T4 displays a tail toward longer 16/100 and shorter 28/141 distances, reflecting occasional compression along the 28/141 direction.
Three conformational classes were defined by the following Cα/Cα distance thresholds:d(16/100)≤ 24 Å and d(28/141) ≥35 Å–16/100‐compressedd(16/100) ≥28 Å and d(28/141)≤ 31 Å–28/141‐compressed25 Å≤ d(16/100) ≤ 27 Å and 32 Å≤ d(28/141)≤34 Å–native‐like.
Criterion 1 was applied first; frames not satisfying it were tested for criterion 2; remaining frames were tested for criterion 3. Frames meeting none of the criteria were left unassigned.
To combine information from both filtering approaches, we retained frames passing both criteria simultaneously. The first approach is DEER‐based and is sensitive to both protein backbone and spin label dynamics. The second relies solely on Cα–Cαdistances and therefore isolates the protein backbone contribution. Three final conformational groups were defined from these intersections:Ellipsoid 1: DEER cluster 1 ∩ Cαcluster 1 (Ellipsoid 1, frames from T2)Ellipsoid 2: DEER cluster 2 ∩ Cαcluster 2 (Ellipsoid 2, frames from T4)Circular: DEER cluster 2 ∩ Cαcluster 3 (Circular, frames from T1).
The Ellipsoid 1 frames come from T2, which demonstrates the same trend in the averaged simulated DEER distributions as the experimental data recorded at 3 kbar. This conformation therefore represents a geometry appearing, but not necessarily predominant, at elevated pressure. The Ellipsoid 2 and Circular frames come from T4 and T1, respectively, both of which reproduce the peak positions observed experimentally at 0 bar. These two conformations thus represent geometries present under normal conditions, with Circular being the predominant state. The three conformational groups were used for all subsequent structural analyses, including pore geometry (HOLE2) and ATP transport energy calculations (Rosetta LigandPathFinder).
Analysis of theVDAC1pore geometry withHOLE2
To characterize the VDAC pore geometry, we used the HOLE2 program (Smart et al.,1996), which calculates the pore radius profile along the channel axis, the minimum pore radius (Rmin), and the estimated macroscopic conductance (Gmacro at 1 M KCl). HOLE2 was applied to every frame from each of the three conformational groups (circular, ellipsoid 1, and ellipsoid 2) using an identical setup.
Atomic radii were assigned using the simple AMBER van der Waals radius set provided with HOLE2. Before analysis, all frames were aligned to a common reference using the backbone heavy atoms of theβ‐barrel in MDAnalysis to ensure a consistent starting orientation. The channel axis was defined along the Z direction, and the starting point for the pore search was set to (39.864, 49.011, 41.263) Å, corresponding to the pore center. A spherical probe was used in all calculations.
Rmin and Gmacro values were visualized as raincloud plots (Figure5b,c). Differences between groups were evaluated using the two‐sided Mann–Whitney U test with Holm correction for multiple comparisons (scipy.stats.mannwhitneyu and statsmodels.stats.multitest.multipletests). Pore radius profiles were interpolated onto a common grid of 500 points spanning the full channel length using linear interpolation, with values outside the sampled region set to NaN. Mean profiles and standard errors of the mean were then calculated for each conformational group (Figure5d). The Python code used for analysis and plotting, together with HOLE2 input files, is available in the Zenodo repository.
Evaluation of the energy ofATPtransport with RosettaLigandPathFinder
To estimate whether ATP transport is hindered in the ellipsoid 1 conformation compared to the native‐like circular conformation, we used the Rosetta LigandPathFinder toolkit. One representative structure from each conformational ensemble was used as input. For ellipsoid 1, we selected the structure with the narrowest bottleneck, defined as the minimum diameter of 12.0 Å between closest heavy atoms (Figure5f). For the circular conformation, we selected the structure closest to the x‐ray structure (PDB:3EMN) by RMSD of β‐barrel backbone heavy atoms.
For Rosetta LigandPathFinder, we used a complex of ATP4−, Mg2+and two water molecules as the ligand. The initial coordinates were taken from DFT calculations, and the library of 275 conformers was created with BCL by employing RMSD clustering at 0.25 Å level (Mendenhall et al.,2021; Mudryk et al.,2020). Guide paths for tunnel exploration were initialized as straight lines going through centers of mass of two structures. Standard parameters were used (full inputs provided in the SI), with the step size of 0.33 Å resulting in trajectories with 153 frames. RMSD restraint for ligand conformations between frames had a standard deviation of 1.0 Å.
For each representative structure, Rosetta LigandPathFinder simulated 100 independent ATP transport trajectories along the VDAC channel axis. Each trajectory was represented as a sequence of ATP coordinate sets and loaded using the ProDy Python library (v2.6.1) (Bakan et al.,2011). Since individual trajectories may differ from one another, we assessed their variability by computing pairwise distances between all trajectory pairs. Each pairwise distance was defined as the uniform‐weight mean of per‐frame RMSDs. Trajectories were then grouped using Affinity Propagation clustering applied to this precomputed distance matrix. For each resulting cluster, the mean interaction energy profile was calculated across all member trajectories. A cluster was classified as dominant if its size exceeded 95% of the size of the largest cluster. The mean energy profile of the dominant cluster was used as the representative profile for each conformational group. Interaction energy profiles were plotted as a function of the channel coordinate (Figure5e). The positioning of ATP in the bottleneck region of the ellipsoid 1 conformation is illustrated in Figure5g,h. Scripts for the analysis and visualization of Rosetta LigandPathFinder results are available in the Zenodo repository.
Additional statistics based on Cα/Cα‐distances‐only filtered frames
To assess pore geometry across a broader and more diverse set of structures, we repeated the HOLE2 analysis on an extended set of frames. In contrast to the combined DEER–Cα/CαDistance approach described above, no DEER‐based filtering was applied here. Frames were selected solely based on Cα/Cαdistance thresholds, and all five trajectories (T1–T5) were included. Each frame was independently assigned to one of three conformational classes if it satisfied the corresponding criteria:d(16/100) ≤24 Å and d(28/141) ≥35 Å–Ellipsoid 1d(16/100) ≥28 Å and d(28/141)≤ 31 Å–Ellipsoid 225 Å≤ d(16/100)≤ 27 Å and 32 Å≤ d(28/141)≤ 34 Å Circular
This classification yieldedn= 75 frames for Ellipsoid 1,n= 131 for Ellipsoid 2, andn= 2026 for Circular, with frames originating from all five trajectories. For each classified frame, HOLE2 was run using the same setup described above. Rmin and Gmacro values were extracted from the HOLE2 output files. Mean pore‐radius profiles and standard errors of the mean were calculated within each conformational class. Rmin and Gmacro distributions were visualized as raincloud plots (SFig.8a,b), and between‐group differences were evaluated using the two‐sided Mann–Whitney U test with Holm correction for multiple comparisons. Pore radius profiles were read from precomputed CSV files and interpolated onto a common grid of 500 points, as described above (Figure5).
The results obtained with this distance‐only classification (T1–T5) are qualitatively and quantitatively similar to those from the combined DEER/distance‐based filtering applied to T1, T2, and T4 (Figure5and SFig.8). However, the DEER‐assisted approach provides clearer and more accurate discrimination between VDAC conformations. The Python code used for this analysis is available in the Zenodo repository.
AUTHOR CONTRIBUTIONS
M. Elgeti:Investigation; funding acquisition; writing – original draft; writing – review and editing; methodology; formal analysis; data curation.L. Bergdoll:Conceptualization; investigation; writing – original draft; formal analysis.A. Zlobin:Methodology; data curation; software.J. P. Duneau:Methodology; software; data curation.J. Belyaeva:Investigation; methodology; formal analysis; data curation.J. Abramson:Conceptualization; funding acquisition; writing – original draft; writing – review and editing; project administration; supervision.W. Hubbell:Supervision; funding acquisition.
FUNDING INFORMATION
This work was supported by NIH grant R35GM135175 (JA), R01GM078844 and the Jules Stein Professorship Endowment (to WLH) and R01GM137081 (to ME). This work was performed using HPC resources from GENCI–IDRIS (Grant 2021‐A0110707044 2022 and 2023‐AD010707044R1).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
References
- AbrahamMJ, MurtolaT, SchulzR, PállS, SmithJC, HessB, et al. GROMACS: high performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX. 2015; 1‐2: 19–25.
- AkasakaK. Highly fluctuating protein structures revealed by variable‐pressure nuclear magnetic resonance. Biochemistry. 2003; 42: 10875–10885. doi.org/10.1021/bi034722p
- AltenbachC. LongDistances—a program to analyze DEER data. EPR Newletter. 2021; 31: 12–13.
- BakanA, MeirelesLM, BaharI. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011; 27: 1575–1577. doi.org/10.1093/bioinformatics/btr168
- BayrhuberM, MeinsT, HabeckM, BeckerS, GillerK, VillingerS, et al. Structure of the human voltage‐dependent anion channel. Proc Natl Acad Sci USA. 2008; 105: 15370–15375. doi.org/10.1073/pnas.0808115105
- BelyaevaJ, ElgetiM. Exploring protein structural ensembles: integration of sparse experimental data from electron paramagnetic resonance spectroscopy with molecular modeling methods. Elife. 2024; 13: e99770. doi.org/10.7554/eLife.99770
- BergdollL, GrabeM, AbramsonJ. An assessment of how VDAC structures have impacted our understanding of their function. In: RostovtsevaTK, editor. Molecular basis for mitochondrial signaling. Cham: Springer International Publishing; 2017. p. 141–160.
- BergdollLA, LerchMT, PatrickJW, BelardoK, AltenbachC, BisignanoP, et al. Protonation state of glutamate 73 regulates the formation of a specific dimeric association of mVDAC1. Proc Natl Acad Sci USA. 2018; 115: E172–E179. doi.org/10.1073/pnas.1715464115
- CasadioR, JacoboniI, MessinaA, de PintoV. A 3D model of the voltage‐dependent anion channel (VDAQ). FEBS Lett. 2002; 520: 1–7. doi.org/10.1016/s0014-5793(02)02758-8
- ChoudharyOP, PazA, AdelmanJL, ColletierJP, AbramsonJ, GrabeM. Structure‐guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat Struct Mol Biol. 2014; 21: 626–632. doi.org/10.1038/nsmb.2841
- ChoudharyOP, UjwalR, KowallisW, CoalsonR, AbramsonJ, GrabeM. The electrostatics of VDAC: implications for selectivity and gating. J Mol Biol. 2010; 396: 580–592. doi.org/10.1016/j.jmb.2009.12.006
- ColombiniM. Voltage gating in the mitochondrial channel, VDAC. J Membr Biol. 1989; 111: 103–111. doi.org/10.1007/BF01871775
- ColombiniM. VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004; 256: 107–115. doi.org/10.1023/b:mcbi.0000009862.17396.8d
- DaniilidisM, GünselU, BroutzakisG, LeitlKD, JanowskiR, FredrikssonK, et al. Structural basis of apoptosis induction by the mitochondrial voltage‐dependent anion channel. Nat Commun. 2025; 16: 9481. doi.org/10.1038/s41467-025-65363-1
- DeardenGI, RavishankarV, SakataKT, MenonAK, BergdollL. Protocol for the production and reconstitution of VDAC1 for functional assays. STAR Protoc. 2024; 5: 103240. doi.org/10.1016/j.xpro.2024.103240
- Gonzalez‐DeWhittKR, ErmolovaN, WangHK, HekstraDR, AlthoffT, AbramsonJ. Insights into VDAC gating: room‐temperature x‐ray Crystal structure of mVDAC‐1. Biomolecules. 2024; 14: 1203. doi.org/10.3390/biom14101203
- HeslopKA, MilesiV, MaldonadoEN. VDAC modulation of cancer metabolism: advances and therapeutic challenges. Front Physiol. 2021; 12: 742839. doi.org/10.3389/fphys.2021.742839
- HillerS, GarcesRG, MaliaTJ, OrekhovVY, ColombiniM, WagnerG. Solution structure of the integral human membrane protein VDAC‐1 in detergent micelles. Science. 2008; 321: 1206–1210. doi.org/10.1126/science.1161302
- HuangJ, RauscherS, NawrockiG, RanT, FeigM, de GrootBL, et al. CHARMM36: an improved force field for folded and intrinsically disordered proteins. Biophys J. 2017; 112: 175a–176a. doi.org/10.1038/nmeth.4067
- LafargueE, DuneauJP, BuzhinskyN, OrnelasP, OrtegaA, RavishankarV, et al. Membrane lipid composition modulates the organization of VDAC1, a mitochondrial gatekeeper. Commun Biol. 2025; 8: 936. doi.org/10.1038/s42003-025-08311-5
- LemastersJJ, HolmuhamedovE. Voltage‐dependent anion channel (VDAC) as mitochondrial governator—thinking outside the box. Biochim Biophys Acta. 2006; 1762: 181–190. doi.org/10.1016/j.bbadis.2005.10.006
- LerchMT, MattRA, MasureelM, ElgetiM, KumarKK, HilgerD, et al. Viewing rare conformations of the beta(2) adrenergic receptor with pressure‐resolved DEER spectroscopy. Proc Natl Acad Sci USA. 2020; 117: 31824–31831. doi.org/10.1073/pnas.2013904117
- LerchMT, YangZ, BrooksEK, HubbellWL. Mapping protein conformational heterogeneity under pressure with site‐directed spin labeling and double electron‐electron resonance. Proc Natl Acad Sci USA. 2014; 111: E1201–E1210. doi.org/10.1073/pnas.1403179111
- MendenhallJ, BrownBP, KothiwaleS, MeilerJ. BCL: : conf: improved open‐source knowledge‐based conformation sampling using the crystallography open database. J Chem Inf Model. 2021; 61: 189–201. doi.org/10.1021/acs.jcim.0c01140
- MertinsB, PsakisG, GrosseW, BackKC, SalisowskiA, ReissP, et al. Flexibility of the N‐terminal mVDAC1 segment controls the channel's gating behavior. PLoS One. 2012; 7: e47938. doi.org/10.1371/journal.pone.0047938
- Michaud‐AgrawalN, DenningEJ, WoolfTB, BecksteinO. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011; 32: 2319–2327. doi.org/10.1002/jcc.21787
- MudrykKD, SeidelR, WinterB, WilkinsonI. The electronic structure of the aqueous permanganate ion: aqueous‐phase energetics and molecular bonding studied using liquid jet photoelectron spectroscopy. Phys Chem Chem Phys. 2020; 22: 20311–20330. doi.org/10.1039/d0cp04033a
- NajbauerEE, Tekwani MovellanK, GillerK, BenzR, BeckerS, GriesingerC, et al. Structure and gating behavior of the human integral membrane protein VDAC1 in a lipid bilayer. J Am Chem Soc. 2022; 144: 2953–2967. doi.org/10.1021/jacs.1c09848
- NgoVA, Queralt‐MartínM, KhanF, BergdollL, AbramsonJ, BezrukovSM, et al. The single residue K12 governs the exceptional voltage sensitivity of mitochondrial voltage‐dependent anion channel gating. J Am Chem Soc. 2022; 144: 14564–14577. doi.org/10.1021/jacs.2c03316
- PannierM, VeitS, GodtA, JeschkeG, SpiessHW. Dead‐time free measurement of dipole–dipole interactions between electron spins. J Magn Reson. 2000; 142: 331–340. doi.org/10.1006/jmre.1999.1944
- PolyhachY, BordignonE, JeschkeG. Rotamer libraries of spin labelled cysteines for protein studies. Phys Chem Chem Phys. 2011; 13: 2356–2366. doi.org/10.1039/c0cp01865a
- QiY, LeeJ, ChengX, ShenR, IslamSM, RouxB, et al. CHARMM‐GUI DEER facilitator for spin‐pair distance distribution calculations and preparation of restrained‐ensemble molecular dynamics simulations. J Comput Chem. 2020; 41: 415–420. doi.org/10.1002/jcc.26032
- Queralt‐MartinM, BergdollL, JacobsD, BezrukovSM, AbramsonJ, RostovtsevaTK. Assessing the role of residue E73 and lipid headgroup charge in VDAC1 voltage gating. Biochim Biophys Acta Bioenerg. 2019; 1860: 22–29. doi.org/10.1016/j.bbabio.2018.11.001
- RosencransWM, KhuntiaH, Ghahari LarimiM, MahalakshmiR, YuTY, BezrukovSM, et al. Conformational plasticity of mitochondrial VDAC2 controls the kinetics of its interaction with cytosolic proteins. Sci Adv. 2025; 11: eadv4410. doi.org/10.1126/sciadv.adv4410
- RostovtsevaT, ColombiniM. ATP flux is controlled by a voltage‐gated channel from the mitochondrial outer membrane. J Biol Chem. 1996; 271: 28006–28008. doi.org/10.1074/jbc.271.45.28006
- RostovtsevaT, ColombiniM. VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J. 1997; 72: 1954–1962. doi.org/10.1016/S0006-3495(97)78841-6
- RostovtsevaTK, KazemiN, WeinrichM, BezrukovSM. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem. 2006; 281: 37496–37506. doi.org/10.1074/jbc.M602548200
- RostovtsevaTK, BezrukovSM. VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr. 2008; 40: 163–170. doi.org/10.1007/s10863-008-9145-y
- SezerD, FreedJH, RouxB. Parametrization, molecular dynamics simulation, and calculation of electron spin resonance spectra of a nitroxide spin label on a polyalanine alpha‐helix. J Phys Chem B. 2008; 112: 5755–5767. doi.org/10.1021/jp711375x
- SmartOS, NeduvelilJG, WangX, WallaceBA, SansomMS. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph. 1996; 14: 354–360, 376. doi.org/10.1016/s0263-7855(97)00009-x
- TeijidoO, RappaportSM, ChamberlinA, NoskovSY, AguilellaVM, RostovtsevaTK, et al. Acidification asymmetrically affects voltage‐dependent anion channel implicating the involvement of salt bridges. J Biol Chem. 2014; 289: 23670–23682. doi.org/10.1074/jbc.M114.576314
- TeijidoO, UjwalR, HillerdalCO, KullmanL, RostovtsevaTK, AbramsonJ. Affixing N‐terminal alpha‐helix to the wall of the voltage‐dependent anion channel does not prevent its voltage gating. J Biol Chem. 2012; 287: 11437–11445. doi.org/10.1074/jbc.M111.314229
- TessmerMH, StollS. chiLife: an open‐source python package for in silico spin labeling and integrative protein modeling. PLoS Comput Biol. 2023; 19: e1010834. doi.org/10.1371/journal.pcbi.1010834
- UjwalR, CascioD, ColletierJP, FahamS, ZhangJ, ToroL, et al. The crystal structure of mouse VDAC1 at 2. 3 a resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA. 2008; 105: 17742–17747. doi.org/10.1073/pnas.0809634105
- VarugheseJT, BuchananSK, PittAS. The role of voltage‐dependent Anion Channel in mitochondrial dysfunction and human disease. Cells. 2021; 10: 1737. doi.org/10.3390/cells10071737
- VermaA, Shteinfer‐KuzmineA, KamenetskyN, PittalaS, PaulA, Nahon CrystalE, et al. Targeting the overexpressed mitochondrial protein VDAC1 in a mouse model of Alzheimer's disease protects against mitochondrial dysfunction and mitigates brain pathology. Transl Neurodegener. 2022; 11: 58. doi.org/10.1186/s40035-022-00329-7
- VirtanenP, OliphantTE, HaberlandM, ReddyT, CournapeauD, BurovskiE, et al. SciPy 1. 0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020; 17: 261–272. doi.org/10.1038/s41592-019-0686-2
- WassenaarTA, PluhackovaK, BockmannRA, MarrinkSJ, TielemanDP. Going backward: a flexible geometric approach to reverse transformation from coarse grained to atomistic models. J Chem Theory Comput. 2014; 10: 676–690. doi.org/10.1021/ct400617g
- YuanS, SunR, ShiH, ChapmanNM, HuH, GuyC, et al. VDAC2 loss elicits tumour destruction and inflammation for cancer therapy. Nature. 2025; 640: 1062–1071. doi.org/10.1038/s41586-025-08732-6
- ZachariaeU, SchneiderR, BrionesR, GattinZ, DemersJP, GillerK, et al. Beta‐barrel mobility underlies closure of the voltage‐dependent anion channel. Structure. 2012; 20: 1540–1549. doi.org/10.1016/j.str.2012.06.015
- ZethK, TheinM. Porins in prokaryotes and eukaryotes: common themes and variations. Biochem J. 2010; 431: 13–22. doi.org/10.1042/BJ20100371
Republished from the open web under CC-BY. Authors: Bergdoll L, Elgeti M, Belyaeva J, Zlobin A, Duneau JP, Hubbell W, Abramson J. Read the original.