How important are quantum mechanical effects in controlling biological functions: Enzymes, electron transfer and bird navigation.
In light of the centennial of the Schrödinger equation, this article addresses the popular idea that quantum mechanical effects play an important role in biological systems. We start by defining what is qualified as a quantum effect. We then clarify the idea that quantum mechanical tunneling is a crucial factor in enzyme catalysis. Here we show that quantum tunneling is in fact anticatalytic and that there is no consistent theoretical and experimental evidence that the tunneling effects are enhanced significantly by enzymes relative to the corresponding reference solution reactions. We next turn to electron transfer reactions, arguing that in most cases the efficiency of the reaction is controlled by classical effects. We also consider the low barrier hydrogen bond idea and clarify its anticatalytic nature. In addition, we present a specific case study of electron transfer in a protein system potentially responsible for bird navigation, providing a concrete example for evaluating the role of quantum and classical effects under biologically relevant conditions. We argue that for the electron transfer step in this system, the most important control is associated with the classical control of the relevant potential surfaces and the fluctuations of the key energy gaps.
WHAT CAN WE LEARN FROM SPECIFIC CASES
Quantum mechanics is the basis of the determination of molecular potential surfaces and the corresponding forces. This fact should be distinguished from the existence of quantum mechanical effects which are effects that cannot be described by classical mechanics. As much as biological molecules are concerned the issue is how important are quantum mechanical effects in biological systems. In addressing this question, we have to recognize the natural inclination to assume that complex biological processes are guided by quantum mechanical factors (e.g., tunneling). This issue is analyzed carefully below.
The role of QM calculations
Before we start considering the possible role of quantum effects in biology, it is useful to consider the undeniable role of quantum mechanical calculations in studies of biological functions. Most notable are QM/MM calculations of enzymatic reactions (Gelfand & Warshel,2025; Kamerlin et al.,2009; Nandi & Warshel,2025) and other processes (Nandi et al.,2024; Senn & Thiel,2009) that have been proven to be crucial for understanding the molecular origin of biological functions. For example, it is basically impossible to determine the origin of the catalytic effect of enzymes without some type of QM/MM calculation (Kamerlin & Warshel,2011). However, this does not mean that we are dealing with quantum mechanical catalytic effects. That is, quantum mechanical calculations are crucial for obtaining potential energy surfaces for molecular processes. However, this does not mean that we are dealing with a quantum mechanical effect. In our view, quantum mechanical effects are effects that cannot be described by classical mechanics treatments of the motion on the corresponding Born Oppenheimer surfaces. In fact, for the purpose of this paper it is not useful to discuss the importance of quantum mechanical effects in biology without defining what we mean by such effects.
It seems to us that effects that can be described by propagating molecular dynamics (MD) trajectories on Born‐Oppenheimer surfaces should not be considered as quantum mechanical effects. In contrast, nuclear quantum effects (NQE) refer specifically to phenomena associated with the quantum nature of nuclear motion that cannot be described by classical mechanics. These include effects such as zero‐point energy and quantum tunneling. In the following sections, we examine whether such effects play a significant functional role in biological systems.
Enzyme catalysis
It has been very appealing to suggest that enzyme catalysis is associated with quantum effects (Marais et al.,2018). Such problematic implications are usually done by invoking terms like “coupling,” “tunneling,” “Coherence,” and more. However, without any successful demonstration that the alleged quantum effects actually lead to a specific observed catalytic effect, it has to take such allegations seriously. In the sections below, we will examine some of the most prevalent proposals.
There is no consistent evidence that quantum tunneling effects contribute to enzyme catalysis
One of the most popular arguments that quantum mechanical effects play an important role in biology involves the idea that quantum mechanical tunneling leads to large catalytic effects. Apparently, this is a very appealing idea, as it implies that the enzyme compresses the donor acceptor distance, creating a narrower potential barrier and large tunneling (Bahnson et al.,1997; Ball,2004; Klinman,1989; Luo et al.,1999; Mincer & Schwartz,2003; Sutcliffe & Scrutton,2000). However, our studies (Liu & Warshel,2007) established that the nuclear quantum mechanical (NQM) effects decrease rather than increase due to compression. That is, when the distance between the donor and acceptor is sufficiently compressed, the mixing between the two electronic states makes the adiabatic surface very flat rather than narrow, so that the tunneling effect decreases (see Kamerlin et al.,2010and Figure1).
The analysis of Figure1has important implications for any proposals that attribute a significant role to NQM in enzyme catalysis. Specifically, the factors that increased NQM contributions are found to be anticatalytic. This behavior arises primarily because the rate constant decreases as the donor–acceptor distance increases, whereas the tunneling increases when the donor acceptor distance increases. The observation that a large kinetic isotope effect (KIE) reflects an increase, rather than a decrease, in the donor–acceptor distance is counterintuitive and has therefore been a persistent source of confusion (see, e.g., Kamerlin &`Warshel,2010bfor a detailed discussion). In our view, a central difficulty with the catalytic tunneling proposal is the fact that, despite our demonstration of the fundamentally anticatalytic nature of the original tunneling picture, namely, donor–acceptor compression reduces rather than enhances tunneling. At this point, it may be useful to further expand on the argument that the tunneling proposal is anticatalytic by pointing out that the tunneling would increase when the donor acceptor distance is forced to increases. At this situation, the rate constant decreases since the barrier becomes higher despite the tunneling correction. The above issues have not been addressed by proponents of catalytic tunneling and a reasonable response is unlikely to emerge.
Kohen and coworkers (Kohen et al.,1999) observed that the activation enthalpy (ΔH‡) for the reaction catalyzed by thermophilic alcohol dehydrogenase (ADH) decreases from 23.6 kcal mol−1at lower temperatures (0–30°C) to 14.6 kcal mol−1at higher temperatures (30–65°C). They interpreted this behavior as evidence for a contribution to kcatarising from vibrationally enhanced tunneling at elevated temperatures. Although this interpretation emphasized dynamical effects, we have demonstrated (Liu & Warshel,2007) that a substantial portion of the observed behavior—and clearly its dominant classical contribution—can be understood as an entropic effect, which can be rationalized by considering changes in the interactions between the reacting solute and its environment associated with variations in the polarity of the reacting atoms. Our analysis further indicates that the observed temperature dependence primarily reflects changes in the donor–acceptor distance. Consequently, the temperature trend implies that tunneling contributions are anticatalytic rather than catalytic (Liu & Warshel,2007).
Overall, the arguments presented in Figure1have not been disputed by proponents of the catalytic tunneling proposal. Moreover, we are not aware of any case in which the tunneling effect simulated or measured in an enzyme significantly exceeds the corresponding effect in the reference solution reaction. Thus there is no real support for the use of tunneling in enzyme catalysis. Of course, to remove any confusion, we are not saying that enzyme reactions do not involve tunneling. What we are saying is that tunneling does not help with enzyme catalysis.
Mode coupling
It has been very popular to suggest that enzymes work by mode coupling (Hammes‐Schiffer,2004; Rod et al.,2003; Watney et al.,2003). Basically, this idea cannot have merit for enzyme catalysis, since the reference reaction in water involves solvent motions that can also be described as reactive modes, and thus the same effect is already present in the reference system, and hence those modes alone cannot contribute to enzymatic catalysis.
More specifically, a general analysis of the coupling between the protein (or solvent) vibrations and the chemical process was introduced by Warshel and coworkers in terms of the dispersed polaron (DP) spin boson treatment (Olsson & Warshel,2004; Warshel & Parson,2001).
The DP approach connects fluctuations of the EVB diabatic energy gap along MD trajectories to those of an equivalent harmonic system. The corresponding power spectrum for fluctuations in a given diabatic state is obtained from the Fourier transform of the energy‐gap autocorrelation function, yielding peaks at frequencies associated with modes coupled to the reaction. In the high‐temperature limit, the amplitudes of these peaks are proportional to the square of the displacement along the corresponding coordinates. Within this framework, it has been possible to reexamine several enzyme systems (Kurian et al.,2016), including some that have been cited as evidence for the importance of rate‐promoting modes in enzyme catalysis (Olsson & Warshel,2004; Warshel & Parson,2001).
The proponents of the mode coupling effects (e.g., Watney et al.,2003) claimed to identify networks of correlated conformational changes with components projected along the reaction path in simulations, but concluded that these correlations actually reflect equilibrium structural effects rather than genuine dynamical contributions. In general, the identification of correlated motions does not introduce any fundamentally new perspective on enzyme catalysis, since solvent reorganization along the reaction coordinate in solution likewise involves strongly correlated structural changes. Importantly, the EVB dispersed‐polaron (DP) analysis provides a consistent means to quantify the projection of protein motions onto the reaction coordinate and allows for a direct, quantitative comparison with the corresponding reference reaction in solution.
In enzymes, coupling between protein motions and the chemical process arises from fluctuating electrostatic interactions between the reacting solute and surrounding charged or polar residues, as well as bound water molecules. In solution, analogous effects originate from the reorientation of the solvation shells. In both cases, the reaction coordinate necessarily contains contributions from environmental (solvent) degrees of freedom. The essential distinction lies in the magnitude of the environmental coordinate changes during the reaction, which determines the reorganization energy and is generally smaller in enzymes due to active‐site preorganization. Consistent with this view, studies employing the DP framework (Kamerlin & Warshel,2010a) have demonstrated that the mode‐coupling picture effectively amounts to expressing the reaction coordinate along a harmonic or quasiharmonic pathway. While this decomposition is useful for analyzing contributions to the reorganization energy, the catalytic effect itself originates from the reduced reorganization energy rather than from any time‐dependent coherence of specific modes.
Entanglement and other problematic proposals
The idea that enzymes work by quantum entanglement can be popular for those who have not tried to truly understand how enzymes work. Such an idea has been proposed for example for the action of type II restriction endonucleases (Kurian et al.,2016). Presuming that the mechanism is unclear. However, the glaring problem with the proposal is that its proponents never tried to reproduce any observed parameters by their model or more importantly to see whether energy based models face any problem in reproducing the observed effects. For example, one should have examined if a reasonable free energy landscape, probably with a kinetic equation, can reproduce the observed time dependence. Using arbitrary claims is not likely to be scientifically useful. Otherwise, one can suggest that the action of restriction enzymes is modulated by gravitational waves. The actual proposal involves an arbitrary phenological Hamiltonian that is not based on any clear energy consideration.
An equally problematic proposal has involved the strange speculation that epigenetics involves quantum effects (Siebert et al.,2023). It is regrettable that such proposals are not met by more serious scrutiny with the requirement of reproducing some observed effects.
Low‐barrier hydrogen bond (LBHB) and covalent catalysis
A widely discussed proposal for the origin of enzyme catalysis is the low‐barrier hydrogen bond (LBHB) hypothesis (Cassidy et al.,1997; Cleland & Kreevoy,1994; Frey et al.,1994; Pan & McAllister,1997). This proposal is based on the assumption that the formation of a partially covalent interaction between enzyme hydrogen bonds and the transition state of the reacting system leads to catalytic enhancement (Cassidy et al.,1997; Pan & McAllister,1997). Because it invokes direct bonding interactions with the enzyme, the LBHB hypothesis may be viewed as an appeal to a quantum mechanical effect. However, this proposal has not been evaluated using rigorous energetic considerations. The primary distinction between the LBHB picture and the well‐established view that preorganized hydrogen bonds stabilize the transition state electrostatically (Warshel & Aqvist,1991) is that an LBHB is assumed to involve a partially covalent, delocalized bond, such as a bond of the form X–H–Y, where Y may represent, for example, a negatively charged oxygen atom of the solute in the transition state (see also Figure2).
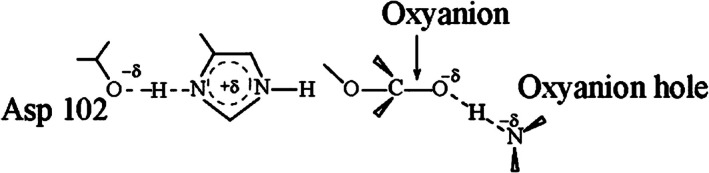
Schematic illustration of the low‐barrier hydrogen bond (LBHB) proposal in serine proteases. The figure depicts a covalent interaction model—whose validity remains to be established—in which active‐site groups, such as Asp102 and the oxyanion hole, form a partial covalent bond with the transition state of the reacting system, both in the enzyme and in solution. Incorporation of environmental effects into the energies of the diabatic states enables a quantitative assessment of how changes in the surroundings influence the charge‐transfer (CT) character of the hydrogen bond. This EVB‐based approach has been applied in quantitative analyses of putative hydrogen bonds to the environment (Garcia‐Viloca et al.,2001; Mulholland et al.,2000) and has shown that the LBHB hypothesis cannot account for the catalytic effect of preorganized hydrogen bonds.
Warshel and Papazyan (1996) demonstrated that the formation of such an LBHB would weaken, rather than enhance, transition‐state solvation and would therefore exert an anticatalytic effect. In contrast, enzymes and polar solutions are more effective at stabilizing the transition state through localized charge interactions than through delocalized bonding interactions (Garcia‐Viloca et al.,2001). It is also important to emphasize that gas‐phase calculations that have been cited in support of the LBHB proposal (Warshel & Aqvist,1991) are not relevant to enzyme active sites, where environmental effects dominate. All available EVB studies (see discussion in [Schutz & Warshel,2004] as well as molecular‐orbital QM/MM studies that reach a quantitatively reliable level [Garcia‐Viloca et al.,2001; Molina et al.,2003; Mulholland et al.,2000]) contradict the LBHB hypothesis.
It is worth emphasizing at this point that, contrary to some implications in the literature (e.g., Weinhold,1997), our analysis of LBHB (Warshel et al.,1995) is based on the empirical valence bond (EVB) framework, which is arguably the most reliable approach currently for examining environmental effects on covalent interactions and charge‐transfer contributions in hydrogen bonding. Within the EVB formulation, the diabatic states and covalent mixing elements are parameterized to reproduce ab initio ground‐state potential energy surfaces and the associated changes in charge distribution observed during gas‐phase reactions.
Electron transfer in proteins
An interesting issue that may confuse the argument about quantum mechanical effects is the issue of biological electron transfer (ET). In this case we would like to know how much ET is accelerated in biological systems relative to the same process in nonbiological media.
Here we start by considering the rate of ET. That is, as was shown in our initial work (Warshel,1982) and subsequent works (Hwang & Warshel,1985; Warshel & Hwang,1986), one can obtain the rate of ET by using a semiclassical surface hopping approach. This involves running classical trajectories on the surface of the reactant and/or combination of the reactant and the product surfaces. While the surface hopping approach involves significant approximations, we find that at least for harmonic potential surfaces in the high temperature limit, the semiclassical expression converges to the exact quantum mechanical expression (Marcus & Sutin,1985). In this limit, the saddle‐point approximation gives:
Hereis the difference between the mean potential energies of the reactant and product states (⟨ϵ2⟩2− ⟨ϵ1⟩1), and λ, the reorganization energy, is the energy required to change the system between the most probable configurations of the two states. From Equation (1) we find that the activation energy of the reaction () is:
If we define the reaction coordinatexas the energy difference (x≡ Δϵ12= ϵ2− ϵ1) and assume that the energy surfaces ϵ1and ϵ2are harmonic functions of this coordinate, the reorganization energy can be related to the displacement of the two minima.
Replacing the potential energies Δϵ12,, andby the corresponding free energies (ΔG12,, and) gives the well‐known Marcus equation (Marcus,1956; Marcus & Sutin,1985).
Coming back to the issue of quantum effects in biology, we can start with the exponential factor. Here we can ask how proteins reduce the corresponding activation barriers. In this case, we have found that it is done by reducing the classical reorganization energy as we established for cytochrome c (Churg et al.,1983) and in the case of bacterial reaction centers (Warshel & Parson,2001; Warshel & Schlosser,1981). Thus the corresponding catalytic effect is not associated with a quantum effect. One can argue that in the so‐called inverted Marcus region, where Equation (1) is not valid and one has to consider transitions between vibronic channels (Warshel & Parson,2001). In this case, it may be suggested that the inverted region blocks the very fast relaxation to the initial ground state. However, our study established that the vibronic channels allow a relatively fast relaxation to the ground state (Parson & Warshel,2004b) and that only because the reorganization energy is tuned to be very similar to the free energy difference (see Figure3) we have an optimal quantum yield (Parson & Warshel,2004a).
Another factor that can be examined as a possible quantum effect is the possibility that preexponential electronic coupling,in Equation1, leads to quantum mechanical advantage. Here the opinions are different, where Gray and coworkers (Beratan et al.,1992) have promoted the idea of specific coupling paths, where Dutton and coworkers (Moser et al.,1992) have promoted a uniform distance dependance of coupling. Here again we have to consider the fact that the reference reaction in water also involves an electronic coupling.
Another direction where one may look for quantum mechanical effects is the limit where the surface hopping approach becomes invalid and one should move to a density matrix treatment. A density matrix approach based on actual microscopic simulations has been developed by Parson and Warshel (2004a,2004b) and applied to bacterial reaction centers (Parson & Warshel,2004a). This study, which is only relevant to very low temperatures, established that the ET in bacterial photosynthesis occurs before the excitation energy uses vibrational relaxations to decay to the minimum of the first excited state. This effect may be classified as a quantum mechanical non‐Marcus effect. However, this effect is not relevant at room temperature.
It is worth noting that in the Marcus inverted regime, quantum mechanical tunneling between vibronic states can, in principle, contribute to an acceleration of ET (Warshel,1980). However, this situation is distinct from the cases of hydride or proton transfer discussed above, where the dominant catalytic effect arises from reduction of the activation barrier. In those systems, conditions that enhance tunneling are generally associated with higher barriers and therefore do not contribute to catalysis.
Antennas of reaction centers
An interesting case where long‐lived quantum coherence is likely to play a functional role in the antenna systems of photosynthetic reaction centers has been suggested by Fleming and coworkers (Ishizaki & Fleming,2012) and others (Engel et al.,2007). This idea has been supported by experimental observations of short‐lived coherence. However, the biological relevance of such observations is far from clear.
First, antenna complexes operate in a warm, wet, and noisy biological environment. Thermal motions, protein vibrations, and solvent interactions lead to rapid decoherence, typically on femtosecond timescales. This makes sustained phase coherence across chromophores extremely difficult to maintain. Second, antenna pigments are strongly coupled to their surrounding protein scaffold and vibrational modes (phonons). This strong environmental coupling continuously disrupts phase relationships, causing energy transport to behave effectively as classical hopping rather than coherent wave‐like motion. Third, energetic disorder—both static and dynamic—dominates real antenna systems. Site‐energy fluctuations localize excitons and prevent the phase matching required for coherent transport across the antenna.
Importantly, near‐unity energy transfer efficiency can already be explained by classical models such as Förster resonance energy transfer and related open‐system approaches. Since these mechanisms are robust across temperatures and mutations, there is little evolutionary pressure for biology to rely on fragile quantum coherence.
While ultrafast spectroscopy does detect coherent oscillations, these signals are often better interpreted as vibronic coherence rather than functionally relevant electronic quantum coherence. In this sense, coherence may be an epiphenomenon of molecular structure rather than a biological design principle.
Finally, reaction center antenna systems appear optimized for robustness and reliability, not delicacy. In fact, environmental noise can enhance transport by preventing exciton trapping, a phenomenon sometimes described as environment‐assisted transport (Rebentrost et al.,2009). Crucially, this enhancement relies on the rapid suppression of coherence rather than its preservation. At any rate, the efficiency of photosynthesis is determined by the charge separation steps and not by the efficiency of the light absorption.
Vision
The primary event in vision involves an extremely efficient surface crossing with a quantum yield that approaches 1.0 (Warshel,1976). Here it is tempting to invoke the involvement of quantum effects.
In analyzing this idea we introduced the first MD simulation of a biological process and the use of semiclassical surface hopping approach in biology (Warshel,1976), This simulation predicted correctly the time scale and efficiency of the primary event. This study and subsequent works led to the realization that the effective surface crossing is associated with conical intersection (Olivucci et al.,1993; Warshel & Chu,2001). Such effect can of course also occur in the reference solution process, but the relevant energy gap is far too large in solution. The protein, on the other hand, tunes the energy gap to a region where the surface crossing is optimized. However, as we have shown in (Schulten et al.,1978), the protein does not generate a perfect permanent conical intersection since its thermal fluctuations modulate the energy gap. Thus we are clearly not dealing with a quantum coherence. It seems to us that the main effect of the protein is to tune the energy gap by using electrostatic effects, not by quantum mechanical effects.
BIRD NAVIGATION AS AN EXAMPLE
Introduction
Magnetoreception in migratory birds represents one of the most intriguing unsolved problems in sensory biology. Unlike vision (Wald,1968; Warshel,1976), olfaction (Buck & Axel,1991), or mechanosensation (Ranade et al.,2015), whose molecular mechanisms are largely established, the biophysical origin of magnetic field detection remains under active investigation (Fleissner et al.,2007; Kirschvink et al.,2001; Schulten et al.,1978; Wiltschko & Wiltschko,2005, Luo et al.,2024). Among the proposed mechanisms, the radical pair model (Ritz et al.,2000) has received substantial experimental and theoretical support, particularly through the work of Hore and co‐workers (Xu et al.,2021), who identified a cryptochrome ofErithacus rubecula(ErCry4a) as a potential magnetosensitive candidate. In this model, as shown in Figure4, photoexcitation of the flavin adenine dinucleotide (FAD) cofactor initiates a sequence of ET steps along a conserved tryptophan chain, generating a spin‐correlated radical pair whose singlet–triplet interconversion can be influenced by weak magnetic fields (Hore & Mouritsen,2016). The dim light present during the night might appear insufficient to modulate the behavior of nocturnal birds; however, moonlight spans a broad visible spectrum with decent intensity (Kieffer & Stone,2005), making it capable of exciting the flavin chromophore. In addition, studies in small songbirds have shown that moonlight intensity correlates with their migratory behavior (Prinz et al.,2025).
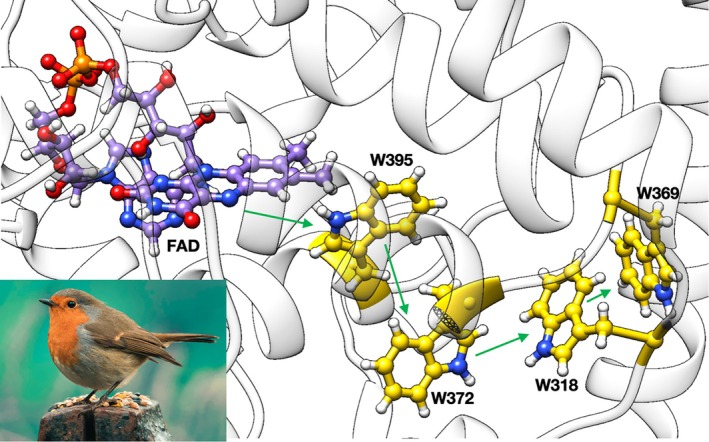
Structure of key residues and cofactors in ErCry4a. The flavin cofactor is in purple and the conserved tryptophan residues forming the ET chain are in gold, with green arrows indicating the photo‐induced electron transfer from FAD and the subsequent charge migration along the tryptophan chain. Inset:Erithacus rubecula(European robin).
Extensive theoretical efforts have been devoted to modeling both the ET energetics and the subsequent spin dynamics. QM/MM (Kattnig & Hore,2017; Luo et al.,2025; Solov'yov et al.,2007) calculations have been employed to characterize charge localization and electronic couplings, while spin Hamiltonian and density‐matrix treatments (Hore & Mouritsen,2016; Timmel et al.,1998) have been used to examine magnetic field effects under realistic hyperfine interactions. Despite these advances, the degree to which the elementary ET steps themselves rely on intrinsically quantum mechanical effects, beyond those associated with spin evolution, remains unclear.
Importantly, while the overall magnetoreception mechanism remains controversial, due to unresolved issues such as the identity of the signaling species, the detailed kinetics of the process, and the extent to which spin coherence can persist under physiological conditions (Hore & Mouritsen,2016; Xu et al.,2021), the second and third ET steps along the tryptophan chain are comparatively well characterized experimentally (Timmer et al.,2023). Their kinetic timescales have been estimated from spectroscopic measurements, providing benchmarkable observables for theoretical analysis. These steps occur on sub‐nanosecond timescales following the initial photoinduced charge separation and are therefore critical in defining the lifetime and stability of the radical pair intermediate.
The availability of experimentally observed ET steps provides an opportunity to examine a more specific question: to what extent are these reactions governed by classical thermally activated processes versus delicate quantum mechanical effects associated with electronic coherence or nonadiabatic dynamics? In the present work, we analyze these two ET steps using EVB simulations combined with diabatic electronic coupling calculations, with the aim of assessing the validity of Marcus‐type behavior, the role of environmental linear response, and the robustness of the process under near‐ambient conditions.
Methods
Initial PDB structure preparation
The protein structure was obtained by using sequence from (Xu et al.,2021) and predicted by AlphaFold Server (Jumper et al.,2021; Varadi et al.,2022). The predicted structure was superimposed with experimentally determined cryptochrome4 (PDB ID:6PU0) (Zoltowski et al.,2019) for visual check and flavin cofactor binding pose determination.
Ab‐initio calculation
The atomic charges for relevant molecular fragments were evaluated Electrostatic Potential (ESP)‐derived charges (Breneman & Wiberg,1990), generated by Gaussian16 (Frisch et al.,2016) at the ωB97X‐D/6–311 + G(d,p) level (Chai & Head‐Gordon,2008) with the Solvation Model based on Density (SMD) (Marenich et al.,2009). The restrained electrostatic potential (RESP)‐fitted charges (Bayly et al.,1993) for each fragment were then generated using AmberTools23 (Case et al.,2023). For these calculations, the tryptophan residue is truncated to beta carbon and cleaved bond saturated with a hydrogen atom. The flavin cofactor is truncated to lumiflavin and the side chain for the ab‐initio calculation.
Electronic couplings between charge‐localized diabatic states were computed using the constrained density functional theory configuration interaction (CDFT‐CI) (Kaduk et al.,2012) approach as implemented in Q‐Chem 6.2 (Epifanovsky et al.,2021). All calculations were performed at a fixed nuclear geometry using the range‐separated hybrid functional ωB97X‐D and the cc‐pVDZ basis set. Solvent effects were included via the integral equation formalism polarizable continuum model (IEF‐PCM) (Cancès et al.,1997) with the following dielectric parameters: static dielectric constant ε = 40 and optical dielectric constant ε∞= 1.76.
In a first step, a constrained DFT (CDFT) (Wu & Van Voorhis,2006) single‐point calculation was carried out to generate a solvent reaction field equilibrated to a specific diabatic charge‐localized state. A spin‐resolved charge constraint was imposed to localize one excess positive charge on the donor fragment, while the total system charge and spin multiplicity were fixed to represent an open‐shell radical cation. The converged PCM reaction field corresponding to this diabatic electronic state was saved and subsequently reused.
In a second step, CDFT‐CI calculations were performed using the frozen solvent reaction field obtained from the initial CDFT calculation, thereby eliminating contributions from solvent reorganization to the electronic coupling. Two diabatic states were constructed by imposing equivalent spin‐resolved charge constraints on the donor and acceptor fragments, respectively. Configuration interaction was then carried out in the subspace spanned by these diabatic states to obtain the diabatic Hamiltonian matrix, from which the electronic coupling was extracted as the off‐diagonal Hamiltonian element. This protocol ensures that the reported electronic couplings correspond to purely electronic interactions between charge‐localized states under a consistent solvent polarization.
Monte Carlo sampling
The term ionization configuration (IC) is defined as the most probable protonation states of ionizable residues in a protein at a certain pH (Nandi et al.,2024). The IC of the studied protein system was obtained by combining intrinsic pKa calculation with PDLD/S‐LRA method and a Monte Carlo procedure (Sham et al.,1997; Singh & Warshel,2010) for IC ensemble free energy sampling by:
wheredenotes the free energy change of the protein transition from a hypothetical reference state in which all amino acids have a zero net charge, to the mth IC.denotes the net charge of the ith residue in the mth IC.denotes the intrinsic pKa of the ith residue calculated by the PDLD/S‐LRA method (Sham et al.,1997).denotes the distance between the ith and the jth residue.denotes a distance dependent dielectric constant to account for charge–charge interaction, defined as:
was sampled by Monte Carlo sampling and the ensemble averaged net charge of each residue can be estimated by:
PDLD/S‐LRA method
The ET free energy between the donor and the acceptor was calculated by the following thermodynamic cycle presented in Figure5. The solvation free energy differences were calculated by the PDLD method within the LRA regime (Sham et al.,2000), and the free energy of aqueous redox reactions should in principle be obtained by experimental measurement or ab initio calculations. However, in the present study, the contributions of aqueous reactions cancel since the ET happens between two tryptophan residues.
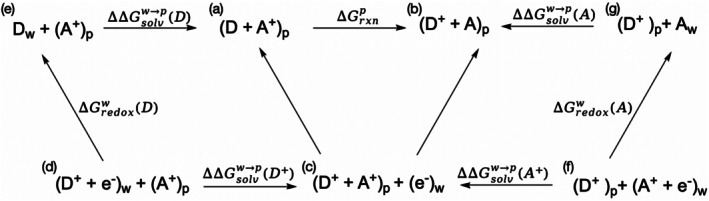
Thermodynamic cycle for evaluation of electron transfer free energy in the protein environment. D and A refer to electron donor and acceptor, respectively, with subscript to indicate the environment (w: bulk water, p: protein). (e−)wdenotes a hypothetical electron donor reservoir in the bulk water but does not directly interact with the system of our interest. The free energies terms calculated are presented on the arrows connecting different states and consistent with the arrow directions. The target reaction is from state (a) to state (b) with free energy change of∆Grxnp.∆∆Gsolvw→pterms refer to the corresponding solvation free energy differences by moving the fragment from water to the protein environment.∆Gredoxwterms refer to the corresponding redox free energy in bulk water. By combining the aqueous redox terms with the corresponding solvation free energy differences, the protein‐phase reaction free energy is obtained. In the present system, where ET occurs between two tryptophan residues, the intrinsic aqueous redox contributions cancel, and∆Grxnpis determined primarily by environmental polarization effects.
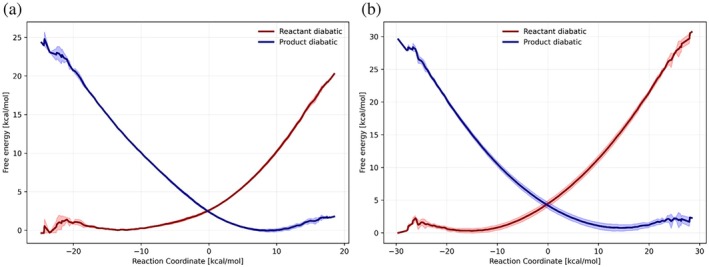
EVB calculations of the electron transfer reactions in the tryptophan chains. (a) and (b) describe the second and the third electron transfer, respectively. Each figure describes the free energy functionals of the corresponding diabatic states (reactant state in red, and product state in blue). Solid lines represent the average free energy values and the corresponding shaded areas refer to the standard deviation.
In order to calculate the ET free energy inside the protein environment, four PDLD calculations are needed to obtain the solvation free energy of each residue in the corresponding redox state. The in‐water properties can either be calculated by high level ab initio or from experiments. In the case of the present study, the in‐water contributions cancel since the ET is between two tryptophans.
The initial phase configuration and momentum of the systems were from the corresponding MD relaxations, with all ionizable residues assigned half of their ensemble averaged charge calculated by the MC procedure (Sham et al.,1997; Singh & Warshel,2010). The electrostatic component of the free energy difference was calculated by averaging 20 configurations sampled in a 20 ps simulation with an effective protein dielectric constant εp= 20 to account for the reorganization effect that is implicit in the semi‐microscopic treatment (Schutz & Warshel,2001; Sham et al.,1998; Singh & Warshel,2010).
EVB method
The EVB method was used to get the corresponding reaction free energy profiles using the MOLARIS‐XG package (Warshel et al.,2012). In these simulations, the reactive region (region 1) includes the donor and acceptor tryptophans, and the remaining protein‐solvent system was assigned to region 2. The system was solvated in a water sphere of radius 20 Å surrounded by Langevin dipoles up to 22 Å and simulated under Surface Constrained All Atom Solvent (SCAAS) boundary conditions (Warshel & King,1985), with long‐range electrostatics treated using the local reaction field approach (Lee & Warshel,1992).
The system was first equilibrated in their reactant state starting from 5.0 K gradually to 274.15 K in a 40 ps simulation with time step size of 1fs, and then another 40 ps simulation on the target temperature has been implemented for the initial phase space sampling of replica for the EVB simulations. The EVB simulations were started from the five independent replicas from relaxation. Each EVB simulation consisted of 31 frames with 10 ps simulation per umbrella sampling window. The EVB Hamiltonian parameters were then obtained by matching the reaction free energies calculated in the thermodynamic cycle by the PDLD/S‐LRA‐2000 method (Sham et al.,2000).
Results
Electronic coupling sensitivity
The electronic coupling terms in Marcus formula were obtained by CDFT‐CI calculations as shown in Table1. Since the protein environment is highly heterogeneous, the CDFT‐CI calculations were done in IEF‐PCM implicit solvation model with various dielectric constants to check the sensitivity of electronic coupling to the polarizability of the environment. It is observed that the electronic coupling is more sensitive to which diabatic electronic state the solvent is interacting with than to the polarizability of the environment. For such reason, we used the electronic couplings calculated with the dielectric constant of 40 for the following Marcus theory calculations.
Table: Electronic coupling of the second electron transfer from CDFT‐CIa.
EVB free energy profile
Our analysis with careful EVB calculations of the second and the third ET reactions (i.e., ET from Trp372 to Trp395, and from Trp318 to Trp372). The results are summarized in Table2and shown in Figure5. The protonation configurations at pH 7.0 and the detailed electron‐transfer free energies from the PDLD calculations are summarized in TablesS1–S3. The corresponding ET rate constants were calculated by Marcus theory (Equation1). The corresponding reorganization energies and activation free energies were directly from EVB profiles, and the electronic coupling is obtained using CDFT‐CI calculations averaged over the reaction field equilibrated in the reactant and product diabatic states.
Table: Calculated Marcus parabolas of the electron transfer reactions and the corresponding observed resultsa.
Discussion
The present study examined the second and third ET steps along the tryptophan chain using a combination of EVB free energy simulations and diabatic electronic coupling calculations. Both reactions are experimentally benchmarkable and occur on sub‐nanosecond timescales, making them among the fastest processes in the system aside from the initial photoinduced charge separation. The calculated free energy profiles exhibit near‐parabolic behavior and yield activation barriers and characteristic times in good agreement with experiment. This agreement indicates that an ensemble‐based free energy description on Born–Oppenheimer potential surfaces is sufficient to capture the essential thermodynamics and kinetics of these steps.
The EVB results further suggest that the solvent and protein environment follow linear response behavior. The forward and backward reorganization energies are comparable in magnitude, and the free energy surfaces are well described by the Marcus formalism. Within this framework, the transition region corresponds to an intermediate polarization state located approximately midway between the reactant and product minima. This observation is consistent with the validity of the linear response approximation under the near‐ambient temperature conditions considered here (274.15 K).
Electronic coupling calculations performed under solvent reaction fields equilibrated separately to the reactant and product diabatic states reveal significant differences between these polarization limits. However, the arithmetic mean of the two limiting couplings yields rate constants that are in good agreement with experiment. This behavior is naturally explained within the Marcus picture: if the solvent response is approximately linear and the coupling varies smoothly with the reaction field, the physically relevant coupling at the crossing region is expected to lie between the two limiting values. The success of the averaged coupling therefore reflects the smooth dependence of the electronic interaction on environmental polarization, rather than requiring explicit trajectory‐based nonadiabatic simulations in which the solvent polarization must be assigned to a partially mixed electronic state along the trajectory. In polarizable environments, determining a physically consistent reaction field corresponding to a coherent superposition of diabatic states is formally ambiguous and often requires additional decoherence corrections. The present approach avoids this ambiguity by evaluating the coupling under well‐defined polarization limits associated with fully localized diabatic states. Importantly, the coupling shows only weak sensitivity to the magnitude of the dielectric constant, indicating that permanent electrostatic polarization, rather than fine‐tuned dielectric response, governs its variation.
The calculated forward and backward ET rates are of comparable magnitude, implying that reversible charge transfer may occur on timescales similar to those of radical pair spin evolution. Such reversible dynamics would lead to stochastic modulation of the spin Hamiltonian through fluctuations in exchange, dipolar, and hyperfine interactions associated with the changing charge distribution. From the perspective of open quantum systems, time‐dependent modulation of the Hamiltonian by nuclear motion is a well‐established source of dephasing. Thus, even without invoking additional mechanisms, thermally driven charge fluctuations may contribute to limiting electronic spin coherence under near‐ambient conditions. A quantitative assessment of this effect would require explicit spin‐dynamics simulations coupled to stochastic ET kinetics and is beyond the scope of the present study.
Taken together, the results indicate that the elementary ET steps in the tryptophan chain operate within a classical Marcus regime characterized by thermally activated barrier crossing, near‐Gaussian environmental response, and smoothly varying electronic interactions. No evidence is found for fragile or finely tuned quantum mechanical control of the charge transfer process itself. If magnetoreception arises from radical pair spin dynamics within this framework, its functional behavior must therefore emerge from collective spin evolution embedded in a thermally fluctuating protein environment, rather than from delicate quantum fine‐tuning at the level of individual molecular interactions.
CONCLUDING REMARKS
The cases considered here indicate that quantum mechanical effects do not play a major role in biology.
We start by pointing out that enzyme catalysis does not involve quantum mechanical effects, not in the form of tunneling enhanced catalysis nor in the form of more exotic effects.
We also consider the possible involvement of quantum mechanical effects in photosynthesis and vision. Here we conclude that also in such light induced systems, proteins operate their functions at room temperature by classical effects.
AUTHOR CONTRIBUTIONS
Aoxuan Zhang:Conceptualization; investigation; writing – original draft; methodology; validation; visualization; writing – review and editing; formal analysis; data curation.Arieh Warshel:Conceptualization; investigation; funding acquisition; writing – original draft; methodology; validation; visualization; writing – review and editing; software; formal analysis; data curation; supervision; resources.
FUNDING INFORMATION
This work was supported by National Science Foundation Grant MCB‐1707167 and National Institute of Health Grant R35 GM122472.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
ACKNOWLEDGMENTS
We thank the University of Southern California, center of High‐Performance Computing (HPC) for computational resources.
Zhang A, Warshel A. How important are quantum mechanical effects in controlling biological functions: Enzymes, electron transfer and bird navigation. Protein Science. 2026;35(7):e70683. 10.1002/pro.70683
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Associated Data
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Bahnson BJ, Colby TD, Chin JK, Goldstein BM, Klinman JP. A link between protein structure and enzyme catalyzed hydrogen tunneling. Proc Natl Acad Sci USA. 1997;94:12797–12802. doi.org/10.1073/pnas.94.24.12797
- Ball P. Enzymes: by chance, or by design? Nature. 2004;431:396–397. doi.org/10.1038/431396a
- Bayly CI, Cieplak P, Cornell W, Kollman PA. A well‐behaved electrostatic potential based method using charge restraints for deriving atomic charges: the resp model. J Phys Chem. 1993;97:10269–10280.
- Beratan DN, Onuchic JN, Winkler JR, Gray HB. Electron‐tunneling pathways in oroteins. Science. 1992;258:1740–1741. doi.org/10.1126/science.1334572
- Breneman CM, Wiberg KB. Determining atom‐centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J Comput Chem. 1990;11:361–373.
- Buck L, Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell. 1991;65:175–187. doi.org/10.1016/0092-8674(91)90418-x
- Cancès E, Mennucci B, Tomasi J. A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys. 1997;107:3032–3041.
- Case DA, Ben‐Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, Cruzeiro VWD, et al. AmberTools23. University of California, San Francisco. 2023.
- Cassidy CS, Lin J, Frey PA. A new concept for the mechanism of action of chymotrypsin: the role of the low‐barrier hydrogen bond. Biochemistry. 1997;36:4576–4584. doi.org/10.1021/bi962013o
- Chai J‐D, Head‐Gordon M. Long‐range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys. 2008;10:6615–6620. doi.org/10.1039/b810189b
- Churg AK, Weiss RM, Warshel A, Takano T. On the action of cytochrome c: correlating geometry changes upon oxidation with activation energies of electron transfer. J Phys Chem. 1983;87:1683–1694.
- Cleland WW, Kreevoy MM. Low‐barrier hydrogen bonds and enzymic catalysis. Science. 1994;264:1887–1890. doi.org/10.1126/science.8009219
- Engel GS, Calhoun TR, Read EL, Ahn T‐K, Mančal T, Cheng Y‐C, et al. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature. 2007;446:782–786. doi.org/10.1038/nature05678
- Epifanovsky E, Gilbert ATB, Feng X, Lee J, Mao Y, Mardirossian N, et al. Software for the frontiers of quantum chemistry: an overview of developments in the Q‐Chem 5 package. J Chem Phys. 2021;155:084801. doi.org/10.1063/5.0055522
- Fleissner G, Stahl B, Thalau P, Falkenberg G, Fleissner G. A novel concept of Fe‐mineral‐based magnetoreception: histological and physicochemical data from the upper beak of homing pigeons. Naturwissenschaften. 2007;94:631–642. doi.org/10.1007/s00114-007-0236-0
- Frey PA, Whitt SA, Tobin JB. A low‐barrier hydrogen bond in the catalytic triad of serine proteases. Science. 1994;264:1927–1930. doi.org/10.1126/science.7661899
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 16, Revision C.01. Wallingford, CT: Gaussian, Inc.; 2016.
- Garcia‐Viloca M, González‐Lafont À, Lluch JM. A qm/mm study of the racemization of vinylglycolate catalyzed by mandelate racemase enzyme. J Am Chem Soc. 2001;123:709–721. doi.org/10.1021/ja002879o
- Gelfand N, Warshel A. Decoding enzymatic dechlorination with multiscale modeling: mechanistic insights into native haloalkane dehalogenase from xanthobacter autotrophicus and its designed variants. ACS Catal. 2025;15:13657–13666. doi.org/10.1021/acscatal.5c03557
- Hammes‐Schiffer S. Quantum–classical simulation methods for hydrogen transfer in enzymes: a case study of dihydrofolate reductase. Curr Opin Struct Biol. 2004;14:192–201. doi.org/10.1016/j.sbi.2004.03.008
- Hore PJ, Mouritsen H. The radical‐pair mechanism of magnetoreception. Annu Rev Biophys. 2016;45:299–344. doi.org/10.1146/annurev-biophys-032116-094545
- Hwang J‐K, Warshel A. Semiclassical simulations of the spectra of anharmonic molecules; problems and alternatives. Chem Phys Lett. 1985;115:281–285.
- Ishizaki A, Fleming GR. Quantum coherence in photosynthetic light harvesting. Annu Rev Condens Matter Phys. 2012;3:333–361. doi.org/10.1098/rsta.2011.0207
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with alphafold. Nature. 2021;596:583–589. doi.org/10.1038/s41586-021-03819-2
- Kaduk B, Kowalczyk T, Van Voorhis T. Constrained density functional theory. Chem Rev. 2012;112:321–370. doi.org/10.1021/cr200148b
- Kamerlin SCL, Haranczyk M, Warshel A. Progress in ab initio QM/MM free‐energy simulations of electrostatic energies in proteins: accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J Phys Chem B. 2009;113:1253–1272. doi.org/10.1021/jp8071712
- Kamerlin SCL, Mavri J, Warshel A. Examining the case for the effect of barrier compression on tunneling, vibrationally enhanced catalysis, catalytic entropy and related issues. FEBS Lett. 2010;584:2759–2766. doi.org/10.1016/j.febslet.2010.04.062
- Kamerlin SCL, Warshel A. An analysis of all the relevant facts and arguments indicates that enzyme catalysis does not involve large contributions from nuclear tunneling. J Phys Org Chem. 2010b;23:677–684. doi.org/10.1002/poc.1620
- Kamerlin SCL, Warshel A. At the dawn of the 21st century: is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010a;78:1339–1375. doi.org/10.1002/prot.22654
- Kamerlin SCL, Warshel A. Multiscale modeling of biological functions. Phys Chem Chem Phys. 2011;13:10401–10411. doi.org/10.1039/c0cp02823a
- Kattnig DR, Hore PJ. The sensitivity of a radical pair compass magnetoreceptor can be significantly amplified by radical scavengers. Sci Rep. 2017;7:11640. doi.org/10.1038/s41598-017-09914-7
- Kieffer HH, Stone TC. The spectral irradiance of the moon. AJ. 2005;129:2887–2901.
- Kirschvink J, Walker M, Diebel C. Magnetite‐based magnetoreception. Curr Opin Neurobiol. 2001;11(4):462–467. doi.org/10.1016/s0959-4388(00)00235-x
- Klinman JP. Quantum mechanical effects in enzyme‐catalysed hydrogen transfer reactions. Trends Biochem Sci. 1989;14:368–373. doi.org/10.1016/0968-0004(89)90010-8
- Kohen A, Cannio R, Bartolucci S, Klinman JP, Klinman JP. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature. 1999;399:496–499. doi.org/10.1038/20981
- Kurian P, Dunston G, Lindesay J. How quantum entanglement in DNA synchronizes double‐strand breakage by type II restriction endonucleases. J Theor Biol. 2016;391:102–112. doi.org/10.1016/j.jtbi.2015.11.018
- Lee FS, Warshel A. A local reaction field method for fast evaluation of long‐range electrostatic interactions in molecular simulations. J Chem Phys. 1992;97:3100–3107.
- Liu H, Warshel A. Origin of the temperature dependence of isotope effects in enzymatic reactions: the case of dihydrofolate reductase. J Phys Chem B. 2007;111:7852–7861. doi.org/10.1021/jp070938f
- Luo J, Benjamin P, Gerhards L, Hogben HJ, Hore PJ. Orientation of birds in radiofrequency fields in the absence of the earth's magnetic field: a possible test for the radical pair mechanism of magnetoreception. J R Soc Interface. 2024;21:20240133. doi.org/10.1098/rsif.2024.0133
- Luo J, Hungerland J, Solov'yov IA, Subotnik JE, Hammes‐Schiffer S. Protein and solvent reorganization drives radical pair stability in avian cryptochrome 4a. J Am Chem Soc. 2025;147:43934–43945. doi.org/10.1021/jacs.5c15726
- Luo J, Kahn K, Bruice TC. The linear dependence of log(kcat/km) for reduction of NAD+by PhCH2OH on the distance between reactants when catalyzed by horse liver alcohol dehydrogenase and 203 single point mutants. Bioorg Chem. 1999;27:289–296.
- Marais A, Adams B, Ringsmuth AK, Ferretti M, Gruber JM, Hendrikx R, et al. The future of quantum biology. J R Soc Interface. 2018;15:20180640. doi.org/10.1098/rsif.2018.0640
- Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta Rev Bioenerg. 1985;811:265–322.
- Marcus RA. On the theory of oxidation‐reduction reactions involving electron transfer. i. J Chem Phys. 1956;24:966–978.
- Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113:6378–6396. doi.org/10.1021/jp810292n
- Mincer JS, Schwartz SD. A computational method to identify residues important in creating a protein promoting vibration in enzymes. J Phys Chem B. 2003;107:366–371.
- Molina PA, Sikorski RS, Jensen JH. NMR chemical shifts in the low‐pH form of a‐chymotrypsin. A QM/MM and ONIOM‐NMR study. Theor Chem Acc. 2003;109:100–107.
- Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature of biological electron transfer. Nature. 1992;355:796–802. doi.org/10.1038/355796a0
- Mulholland AJ, Lyne PD, Karplus M. Ab initio QM/MM study of the citrate synthase mechanism. A low‐barrier hydrogen bond is not involved. J Am Chem Soc. 2000;122:534–535.
- Nandi A, Warshel A. The action of plastic degrading enzyme is accelerated mainly due to an increase in thermal stability rather than by an inherent catalytic effect. J Am Chem Soc. 2025;147:30447–30454. doi.org/10.1021/jacs.5c10598
- Nandi A, Zhang A, Chu ZT, Xie WJ, Xu Z, Dong S, et al. Exploring the light‐emitting agents in renilla luciferases by an effective QM/MM approach. J Am Chem Soc. 2024;146:13875–13885. doi.org/10.1021/jacs.4c00963
- Olivucci M, Ragazos IN, Bernardi F, Robb MA. A conical intersection mechanism for the photochemistry of butadiene. A MC‐SCF study. J Am Chem Soc. 1993;115:3710–3721.
- Olsson MHM, Warshel A. Solute solvent dynamics and energetics in enzyme catalysis: the sN 2 reaction of dehalogenase as a general benchmark. J Am Chem Soc. 2004;126:15167–15179. doi.org/10.1021/ja047151c
- Pan Y, McAllister MA. Characterization of low‐barrier hydrogen bonds. 5. Microsolvation of enol−enolate. An ab initio and DFT investigation. J Org Chem. 1997;62:8171–8176. doi.org/10.1021/jo971290d
- Parson WW, Warshel A. A density‐matrix model of photosynthetic electron transfer with microscopically estimated vibrational relaxation times. Chem Phys. 2004a;296:201–216.
- Parson WW, Warshel A. Dependence of photosynthetic electron‐transfer kinetics on temperature and energy in a density‐matrix model. J Phys Chem B. 2004b;108:10474–10483.
- Prinz D, Heim RJ, Meinken M, Niemann N, Temme L, Esther A, et al. Lunar cycle and moonlight intensity influence nocturnal migration patterns in a small songbird. Sci Rep. 2025;15:19944. doi.org/10.1038/s41598-025-04270-3
- Ranade SS, Syeda R, Patapoutian A. Mechanically activated ion channels. Neuron. 2015;87:1162–1179. doi.org/10.1016/j.neuron.2015.08.032
- Rebentrost P, Mohseni M, Kassal I, Lloyd S, Aspuru‐Guzik A. Environment‐assisted quantum transport. New J Phys. 2009;11:033003.
- Ritz T, Adem S, Schulten K. A model for photoreceptor‐based magnetoreception in birds. Biophys J. 2000;78:707–718. doi.org/10.1016/S0006-3495(00)76629-X
- Rod TH, Radkiewicz JL, Brooks CL. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc Natl Acad Sci USA. 2003;100:6980–6985. doi.org/10.1073/pnas.1230801100
- Schulten K, Swenberg CE, Weller A. A biomagnetic sensory mechanism based on magnetic field modulated coherent electron spin motion. Z Phys Chem. 1978;111:1–5.
- Schutz CN, Warshel A. The low barrier hydrogen bond (LBHB) proposal revisited: the case of the asp ⋯ his pair in serine proteases. Proteins. 2004;55:711–723. doi.org/10.1002/prot.20096
- Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi.org/10.1002/prot.1106
- Senn HM, Thiel W. QM/MM methods for biomolecular systems. Angew Chem Int Ed. 2009;48:1198–1229. doi.org/10.1002/anie.200802019
- Sham YY, Chu ZT, Tao H, Warshel A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD‐LRA, and PDLD/S‐LRA calculations of ligands binding to an HIV protease. Proteins. 2000;39:393–407.
- Sham YY, Chu ZT, Warshel A. Consistent calculations of pKa's of ionizable residues in proteins: semi‐microscopic and microscopic approaches. J Phys Chem B. 1997;101:4458–4472.
- Sham YY, Muegge I, Warshel A. The effect of protein relaxation on charge‐charge interactions and dielectric constants of proteins. Biophys J. 1998;74:1744–1753. doi.org/10.1016/S0006-3495(98)77885-3
- Siebert R, Ammerpohl O, Rossini M, Herb D, Rau S, Plenio MB, et al. A quantum physics layer of epigenetics: a hypothesis deduced from charge transfer and chirality‐induced spin selectivity of DNA. Clin Epigenetics. 2023;15:145. doi.org/10.1186/s13148-023-01560-3
- Singh N, Warshel A. Absolute binding free energy calculations: on the accuracy of computational scoring of protein–ligand interactions. Proteins. 2010;78:1705–1723. doi.org/10.1002/prot.22687
- Solov'yov IA, Chandler DE, Schulten K. Magnetic field effects in arabidopsis thaliana cryptochrome‐1. Biophys J. 2007;92:2711–2726. doi.org/10.1529/biophysj.106.097139
- Sutcliffe MJ, Scrutton NS. Enzymology takes a quantum leap forward. Philos Trans A Math Phys Eng Sci. 2000;358:367–386. doi.org/10.1098/rsta.2000.0536
- Timmel CR, Till U, Brocklehurst B, Mclauchlan KA, Hore PJ. Effects of weak magnetic fields on free radical recombination reactions. Mol Phys. 1998;95:71–89. doi.org/10.1080/09553000050176270
- Timmer D, Frederiksen A, Lünemann DC, Thomas AR, Xu J, Bartölke R, et al. Tracking the electron transfer cascade in european robin cryptochrome 4 mutants. J Am Chem Soc. 2023;145:11566–11578. doi.org/10.1021/jacs.3c00442
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi.org/10.1093/nar/gkab1061
- Wald G. The molecular basis of visual excitation. Nature. 1968;219:800–807. doi.org/10.1038/219800a0
- Warshel A, Aqvist J. Electrostatic energy and macromolecular function. Annu Rev Biophys Biophys Chem. 1991;20:267–298. doi.org/10.1146/annurev.bb.20.060191.001411
- Warshel A, Chu ZT, Villa J, Strajbl M, Schutz C, Shurki A, et al. Molaris‐Xg, version 9.15. Los Angeles, CA: University of Southern California; 2012.
- Warshel A, Chu ZT. Nature of the surface crossing process in bacteriorhodopsin: computer simulations of the quantum dynamics of the primary photochemical event. J Phys Chem B. 2001;105:9857–9871.
- Warshel A, Hwang J‐K. Simulation of the dynamics of electron transfer reactions in polar solvents: semiclassical trajectories and dispersed polaron approaches. J Chem Phys. 1986;84:4938–4957.
- Warshel A, King G. Polarization constraints in molecular dynamics simulation of aqueous solutions: the surface constraint all atom solvent (SCAAS) model. Chem Phys Lett. 1985;121:124–129.
- Warshel A, Papazyan A, Kollman PA. On low‐barrier hydrogen bonds and enzyme catalysis. Science. 1995;269:102–106. doi.org/10.1126/science.7661987
- Warshel A, Papazyan A. Energy considerations show that low‐barrier hydrogen bonds do not offer a catalytic advantage over ordinary hydrogen bonds. Proc Natl Acad Sci USA. 1996;93:13665–13670. doi.org/10.1073/pnas.93.24.13665
- Warshel A, Schlosser DW. Electrostatic control of the efficiency of light‐induced electron transfer across membranes. Proc Natl Acad Sci USA. 1981;78:5564–5568. doi.org/10.1073/pnas.78.9.5564
- Warshel A. Bicycle‐pedal model for the first step in the vision process. Nature. 1976;260:679–683. doi.org/10.1038/260679a0
- Warshel A. Dynamics of reactions in polar solvents. Semiclassical trajectory studies of electron‐transfer and proton‐transfer reactions. J Phys Chem. 1982;86:2218–2224.
- Warshel A. Role of the chlorophyll dimer in bacterial photosynthesis. Proc Natl Acad Sci USA. 1980;77:3105–3109. doi.org/10.1073/pnas.77.6.3105
- Warshel AW, Parson W. Dynamics of biochemical and biophysical reactions: insight from computer simulations. Q Rev Biophys. 2001;34:563–679. doi.org/10.1017/s0033583501003730
- Watney JB, Agarwal PK, Hammes‐Schiffer S. Effect of mutation on enzyme motion in dihydrofolate reductase. J Am Chem Soc. 2003;125:3745–3750. doi.org/10.1021/ja028487u
- Weinhold F. Nature of h‐bonding in clusters, liquids, and enzymes: an ab initio, natural bond orbital perspective. J Mol Struct (Theochem). 1997;398–399:181–197.
- Wiltschko W, Wiltschko R. Magnetic orientation and magnetoreception in birds and other animals. J Comp Physiol A. 2005;191:675–693. doi.org/10.1007/s00359-005-0627-7
- Wu Q, Van Voorhis T. Constrained density functional theory and its application in long‐range electron transfer. J Chem Theory Comput. 2006;2:765–774. doi.org/10.1021/ct0503163
- Xu J, Jarocha LE, Zollitsch T, Konowalczyk M, Henbest KB, Richert S, et al. Magnetic sensitivity of cryptochrome 4 from a migratory songbird. Nature. 2021;594:535–540. doi.org/10.1038/s41586-021-03618-9
- Zoltowski BD, Chelliah Y, Wickramaratne A, Jarocha L, Karki N, Xu W, et al. Chemical and structural analysis of a photoactive vertebrate cryptochrome from pigeon. Proc Natl Acad Sci USA. 2019;116:19449–19457. doi.org/10.1073/pnas.1907875116
Republished from the open web under CC-BY. Authors: Zhang A, Warshel A. Read the original.