Reductive Methylation: An Alternative to Lysine → Arginine Mutagenesis.
Modification of lysine residues is a common strategy in protein engineering, whether to prevent posttranslational modifications, control bioconjugation, or improve crystallization. The standard genetic approach-replacement with arginine by site-directed mutagenesis-preserves positive charge but alters other physicochemical attributes and cannot address the N-terminal amino group. Here, we characterize reductive methylation as a chemical alternative. This reaction converts every primary amino group to a dimethylamino group rapidly under mild aqueous conditions. Using human ribonuclease 1 and a cytotoxic variant engineered to evade the endogenous ribonuclease inhibitor as model systems, we assess the effects of complete dimethylation on thermostability, enzymatic catalysis, protein-protein interaction, compatibility with bioconjugation, cellular uptake, and intracellular persistence. Dimethylation preserves thermostability and a protein-protein interaction. Enzymatic catalysis, in contrast, is reduced by 10 2 - to 10 3 -fold, consistent with the role of catalytic lysine residues. Dimethylation is fully compatible with bioconjugation chemistry. Dimethylated and unmodified ribonucleases show comparable uptake and persistence in human cells. These findings establish reductive methylation as a practical and conservative strategy for lysine modification in protein and peptide engineering and support its use in applications such as biological proteolysis-targeting chimeras (bioPROTACs).
Introduction
Among the canonical amino acids, none rivals lysine in the chemical diversity of its posttranslational modifications [1,2,3,4,5]. Theε‐amino group serves as the attachment point for methyl [6,7,8], acetyl, and ubiquitin‐like modifiers, while its protonated form mediates the Coulombic interactions and hydrogen bonds that drive protein stability, substrate binding, and enzymatic catalysis [9,10]. This versatility makes lysine a frequent target of protein engineering: researchers modify lysine residues to prevent ubiquitination and extend protein half‐life [11,12,13], to improve crystallization [14,15], or to dissect contributions to function [16,17]. As the field of targeted protein degradation matures and protein‐based degraders (bioPROTACs) move toward intracellular applications [18,19,20,21,22,23,24], the ability to modify lysine residues in a controlled, well‐characterized manner has taken on greater practical importance.
The standard genetic approach to eliminating the reactivity of a lysine residue is its replacement with arginine via site‐directed mutagenesis [25,26,27]. Arginine preserves a positive charge at physiological pH and approximates the side‐chain length of lysine. Nonetheless, the two residues differ in important respects (Figure1). The guanidinium group of arginine has a pKaapproximately 2.8 units higher than the ammonium group of lysine, rendering it essentially permanently protonated under biological conditions [28,29]. The planar, Y‐shaped guanidinium group presents five hydrogen‐bond donors in a geometry distinct from that of the tetrahedral ammonium group of lysine, which has three donors, and its larger van der Waals surface can perturb packing at protein–protein interfaces. Moreover, substitutions can require multiple rounds of mutagenesis, and the approach cannot address the N‐terminalα‐amino group, which is itself a substrate for ubiquitination [11,30] and other modifications.
![Conservative chemical and biological alterations to anl‐lysine residue (Lys). Lys can be converted to aNε,Nε‐dimethyl‐l‐lysine residue (dmLys) by reductive methylation. Lys can be converted to anl‐arginine residue (Arg) by site‐directed mutagenesis. pKaand cLogpvalues are for the conjugate acids of methylamine (Lys) [28], trimethylamine (dmLys) [28], and methylguanidine (Arg) [29]. Side chains are depicted as space‐filling models.](https://pub-4f2af807d45642319f8e7f07a4c88770.r2.dev/PMC13270353/PSC-32-e70110-g006.jpg)
Conservative chemical and biological alterations to anl‐lysine residue (Lys). Lys can be converted to aNε,Nε‐dimethyl‐l‐lysine residue (dmLys) by reductive methylation. Lys can be converted to anl‐arginine residue (Arg) by site‐directed mutagenesis. pKaand cLogpvalues are for the conjugate acids of methylamine (Lys) [28], trimethylamine (dmLys) [28], and methylguanidine (Arg) [29]. Side chains are depicted as space‐filling models.
Reductive methylation offers a chemical alternative that, in principle, is more conservative (Figure1). Indeed, before the advent of routine site‐directed mutagenesis, reductive methylation was the foundational technique for blocking ubiquitination. Reductively methylated substrates and “chain‐terminating” reductively methylated ubiquitin were instrumental in deciphering the ubiquitin‐proteasome pathway [31,32,33,34]. The reaction can converteveryprimary amino group, both theε‐amino groups of lysine side chains and theα‐amino group of the N terminus, to a dimethylamino group through imine formation with formaldehyde, followed by reduction with a mild hydride donor [14,15]. Only two CH2groups (six atoms) are added per site. The resulting dimethylammonium group retains a positive charge at physiological pH, with a pKashift of only −0.8 units relative to the parent ammonium group (Figure1), and it can no longer serve as a substrate for ubiquitin ligases. The reductive methylation reaction proceeds to completion under mild aqueous conditions (4°C, pH 7.5) within hours and has been used as a routine rescue strategy for protein crystallization [15]. Despite its simplicity, reductive methylation has not been characterized systematically, an omission that leaves protein engineers and peptide chemists without quantitative guidance on how the modification affects function.
Here, we use ribonucleases as a model system to fill that gap. Ribonuclease (RNase) 1, a human homolog of bovine pancreatic ribonuclease (RNase A), and QBI‐139, a variant that manifests cytotoxic ribonucleolytic activity [35,36,37,38], together present a rich set of testable properties: both proteins have eight lysine residues and an N terminus available for modification; three lysine residues (Lys7, Lys41, and Lys66) are intimately involved in substrate binding and turnover [17,39,40]; and QBI‐139 engages RI in a protein–protein interaction [37,38]. We characterize the effects of complete dimethylation on thermostability, enzymatic catalysis, affinity for RI, compatibility with orthogonal bioconjugation chemistry, cellular uptake, and intracellular persistence. Our results demonstrate that reductive methylation is conservative for many protein properties with the notable exception of catalysis, where the modification is consequential.
Materials and Methods
Conditions
All procedures were performed at ambient temperature (∼22°C) and pressure (1.0 atm) unless indicated otherwise.
Materials
Commercially available reagents and solvents were reagent grade or better from Sigma–Aldrich (St. Louis, MO), unless specified otherwise, and were used directly without further purification. RNase A (≥70 Kunitz units mg−1) was product #R6513, and BL21(DE3)Escherichia colicells were product #69450‐3 from Sigma‐Aldrich.
A 3.5 kDa MWCO dialysis tubing was from Spectrum Labs (Rancho Dominguez, CA). Amicon 15 mL 10 kDa MWCO centrifugal filters were from Thermo Fisher Scientific (Waltham, MA). Zeba spin desalting columns (0.5 mL, 7 kDa MWCO) were from Thermo Fisher Scientific (product #89882). Clear tissue culture‐treated 96‐well plates with flat‐bottomed wells were from Greiner (Kremsmünster, Austria) (product #655160). CellTiter 96 AQueous One Solution Cell Proliferation Assay was from Promega (Madison, WI) (product #G3580). The 0.5 M sodium phosphate buffer, pH 7.0, was from Thermo Fisher Scientific (product #J63791.AK).
Instruments
E. colicells were lysed with a benchtop cell disruptor from Constant Systems (Daventry, UK). FPLC was performed with an ÅKTA pure FPLC system from Cytiva (Wilmington, DE). Protein concentrations were determined using a DS‐11 UV–vis spectrophotometer from DeNovix (Wilmington, DE) and validated using a BCA assay kit (Thermo Fisher Scientific, product #675801). Absorbance measurements were made with a Spark plate reader from Tecan (Männedorf, Switzerland). Protein ESI mass spectrometry was performed with a 6350 Accurate‐Mass Q‐TOF LC/MS instrument from Agilent Technologies (Santa Clara, CA) equipped with a PLRP‐S column (1000 Å pore size, 5‐μm particle size, 50 mm length × 2.1 mm ID). Q‐TOF LC/MS measurements used a linear gradient (5%–95% v/v) of ACN (0.1% v/v formic acid) in water (0.1% v/v formic acid) over 15 min. Peptides were synthesized with a Liberty Blue automated microwave peptide synthesizer from CEM (Matthews, NC). Peptide mass was determined with a MALDI‐TOF microflex LRF instrument from Bruker (Billerica, MA).
Data were plotted and analyzed with Prism 11.0.0 from GraphPad Software (San Diego, CA). Values of cLogPwere calculated with ChemDraw 20.1.0.112 from PerkinElmer (Shelton, CT).
Production and Purification of Ribonucleases
QBI‐139 was a gift from Dr. L. E. Strong (Quintessence Biosciences). M‐DDDDK‐RNase 1 and A19C/G88R RNase A were produced inE. coliwithout a signal peptide but with an N‐terminal methionine residue essentially as described previously [41,42,43]. M‐DDDDK‐RNase 1 was used to make authentic RNase 1 (i.e., without an N‐terminal methionine residue) after enterokinase cleavage [43]. A19C/G88R RNase A is an RI‐evasive variant with Cys19 installed for conjugation [41].
In C4R/P19C/C118V QBI‐139, the fifth disulfide bond in QBI‐139 was reverted to its wild‐type residues, and Cys19 was installed for conjugation. Expression plasmids were transformed into BL21(DE3)E. colicells. A 50 mL starter culture was inoculated from a single colony and grown overnight at 37°C with constant shaking at 250 rpm in terrific broth (TB) containing ampicillin (200 μg mL−1). 1‐L cultures were initiated from the starter culture at an OD600 nm= 0.05 and grown at 37°C in TB containing ampicillin (200 μg mL−1) with constant shaking at 250 rpm. Gene expression was induced with a final concentration of 1.0 mM isopropyl‐β‐d‐1‐thiogalactopyranoside (IPTG) when cultures reached OD600 nm= 1.8 and were grown for an additional 3 h at 37°C with constant shaking at 250 rpm. Cells were pelleted by centrifugation at 6000gfor 15 min at 4°C, and cell pellets were stored at −80°C until resuspension and lysis.
To reduce any mixed disulfides with Cys19, tris(2‐carboxyethyl)phosphine (TCEP) was added at a fivefold molar excess, and the resulting solution was incubated at 4°C for 1 h. The solution was then loaded onto a HiTrap SP cation‐exchange column in 50 mM sodium acetate buffer, pH 5.0, and C4R/P19C/C118V QBI‐139 was eluted with 1.0 M NaCl. Fractions containing C4R/P19C/C118V QBI‐139 were pooled, and the pH was adjusted to 8.0 with 1.0 M Tris–HCl buffer, pH 8.0. DTNB was added at a fivefold excess from a 5 mM stock solution in 50 mM Tris–HCl buffer, pH 8.0, containing EDTA (10 mM) and NaCl (50 mM). Upon incubation at 4°C for 10 min, the solution turned yellow. The pH was adjusted back to 5.0 with 3.0 M sodium acetate buffer, and the mixture was incubated at 4°C overnight. The resulting sample was further purified using a HiTrap SP cation‐exchange column with a linear gradient of NaCl (0.0–1.0 M) in 50 mM sodium acetate buffer, pH 5.0, over 20 column volumes. Fractions containing C4R/P19C/C118V QBI‐139 were pooled, concentrated, and exchanged into Tris–HCl buffer, pH 7.2, containing 50 mM NaCl using an Amicon 15 mL 10‐kDa MWCO spin concentrator. Protein identity was confirmed with SDS–PAGE and Q‐TOF mass spectrometry. Aliquots were flash‐frozen in N2(l) and stored at −80°C.
Production and Purification of Ribonuclease Inhibitor
Recombinant human RI was produced inE. coliand purified essentially as described previously [44]. Purified RI was dialyzed into 25 mM HEPES–NaOH buffer, pH 7.5, containing NaCl (150 mM), DTT (10 mM), and glycerol (10% v/v), flash‐frozen in N2(l), and stored at −80°C.
Reductive Methylation
Reductive methylation of RNase 1 and QBI‐139 was performed as described previously [15] with modifications. Purified protein was diluted to 1–2 mg mL−1in 50 mM sodium phosphate buffer, pH 7.5. Borane·dimethylamine complex was added from a 1.0 M stock solution at a 10‐fold excess over the number of protein amino groups. Then, formaldehyde was added from a 1.0 M stock in a 20‐fold excess relative to the number of amino groups on the protein. The reaction mixture was incubated for ≥2 h at 4°C. The procedure was repeated to ensure complete labeling. After the second incubation, free glycine was added to a final concentration of 100 mM to sequester the formaldehyde, and the reaction mixture was incubated for 2 h at 4°C. Dimethylated proteins were exchanged into DPBS without Ca2+/Mg2+using an Amicon 0.5 mL 10‐kDa MWCO spin concentrator for cytotoxicity assays or into 50 mM Tris–HCl buffer, pH 7.5, containing NaCl (50 mM) for other assays. Reductively methylated proteins are designated with the prefix “dm”.
Protein Thermostability Assay
The thermostabilities of RNase 1, QBI‐139, and their dimethylated variants were assessed in 100 mM Tris–HCl buffer, pH 7.5, containing NaCl (100 mM) using differential scanning fluorimetry as described previously [45].
Enzymatic Activity Assay
Ribonucleolytic activity was measured in 100 mM Tris–HCl buffer, pH 7.5, containing NaCl (100 mM) using a fluorogenic tetranucleotide substrate, 6‐FAM–dArU (dA)2–6‐TAMRA, as described previously [45].
Preparation of DEF–G88R RNase A
DEF–G88R RNase A was prepared essentially as described previously [41]. Briefly, NTB‐capped A19C/G88R RNase A was deprotected with tris(2‐carboxyethyl) phosphine, and the nascent cysteine wasS‐alkylated with the pH‐sensitive fluorophore, 2′,7′‐diethylfluorescein‐5‐iodoacetamide (DEFIA), which was a gift from Dr. L. D. Lavis (University of Wisconsin–Madison).
Affinity for Ribonuclease Inhibitor
The affinity of QBI‐139 and dmQBI‐139 for RI was measured with a competition assay, as described previously [41]. In this assay, the fluorescence of RI‐bound DEF–G88R RNase A in PBS containing BSA (0.1 mg mL−1) and DTT (10 mM) increases upon dissociation driven by competition with QBI‐139 or dmQBI‐139.
Human Cell Culture
K‐562 human myeloid leukemia cells and HAP1 human near‐haploid leukemia cells were obtained from the Robert A. Swanson (1969) Biotechnology Center at the Koch Institute for Integrative Cancer Research at MIT. HEK 293AAV LgBiT human embryonic kidney cells were a gift from Prof. G. M. Church (Harvard Medical School). K‐562 cells were grown in RPMI‐1640 from Thermo Fisher Scientific (product #11875093). HAP1 cells were grown in IMDM from Thermo Fisher Scientific (product #12440053). HEK 293AAV LgBiT cells were grown in DMEM from Thermo Fisher Scientific (product #11995081). RPMI‐1640, IMDM, and DMEM were supplemented with fetal bovine serum from Corning (product #45001‐108) and a solution of penicillin (104units mL−1)–streptomycin (104μg mL−1) from Thermo Fisher Scientific (product #15140122) to make complete media. Cells were cultured in an incubator maintained at 37°C and humidified to 5% v/v CO2(g). Cells were collected with TrypLE Express (Thermo Fisher Scientific, product #12605010) during passaging. All experiments were performed with cells that had <20 passages and tested negative for mycoplasma using the MycoAlertTM PLUS Assay from Lonza Biosciences (product #LT07‐710) at the Koch Institute for Integrative Cancer Research at MIT.
Human Cell Viability Assay
Cell viability in the presence of QBI‐139 and dmQBI‐139 was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay. The metabolic activity of viable cells was measured through the conversion of 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) into a colored, soluble formazan whose absorbance at 490 nm is directly proportional to the number of viable cells [46]. Cells were seeded at 5000 cells per well in a 96‐well tissue culture‐treated microplate and incubated for 24 h. Cells were then treated with either QBI‐139 or dmQBI‐139. Samples were serially diluted twofold in PBS without Ca2+/Mg2+, starting at 30 μM for QBI‐139 and 60 μM for dmQBI‐139. Each treatment volume did not exceed 10% of the medium volume. After 48 h of treatment, 20 μL of MTS reagent was added to the cells. After incubation for ≥2 h at 37°C, the absorbance was recorded at 490 nm. Values represent data collected in biological triplicates with at least three technical replicates and are normalized to the average background absorbance signal from formazan in the medium alone and from vehicle‐treated cells (<10% PBS). Values of IC50, which are the concentrations of a ribonuclease that give half‐maximal cell viability, were calculated by fitting the data using a four‐parameter variable slope model (log[agonist] versus response). The significance of QBI‐139 and dmQBI‐139 additions was determined with an unpaired, two‐tailedt‐test.
Peptide Synthesis
MVSGWRLFKKIS (HiBiT) and N3‐(CH2)5C(O)‐SGSGVSGWRLFKKIS (azidoHiBiT) were synthesized on a 0.05‐mmol scale on a Rink Amide ProTide resin from CEM (0.59 mmol per gram) and a cycle of 2‐min coupling (90°C), 1‐min deprotection (90°C), and 1‐min associated washes and liquid handling. For azidoHiBiT, the final step was an amide coupling between the N‐terminal serine residue and 6‐azidohexanoic acid from Sigma‐Aldrich. Deprotection was performed with 4‐methylpiperidine (20% v/v) and 1‐hydroxybenzotriazole monohydrate (0.10 M) in DMF. Coupling reactions were performed with 5 equiv. of an Fmoc‐protected amino acid, DIC, and Oxyma in DMF. Peptides were cleaved with 82.5:5:5:2.5:5 TFA/phenol/thioanisole/DODT/H2O and purified by HPLC. Pure fractions were combined and lyophilized to obtain a white powder.
Thiol–Maleimide Conjugation
NTB‐capped C4R/P19C/C118V QBI‐139 was subjected to reductive methylation as described in Section2.6. Cys19 in the unmodified and dimethylated protein was deprotected by the addition of TCEP to a concentration of 5 mM and incubation at 4°C for 15 min. The proteins were exchanged into 50 mM sodium phosphate buffer, pH 7.0, containing EDTA (2 mM) using a Zeba spin desalting column and then incubated for 1 h with a fivefold molar excess of dibenzocyclooctyne‐PEG4‐maleimide, which was from Sigma‐Aldrich (product #760676). Excess maleimide was removed by passing the reaction mixture through a Zeba spin desalting column, exchanging into DPBS without Ca2+/Mg2+. The final protein concentration was determined by measuring the UV absorption at 280 nm.
Semisynthesis of HiBiT Conjugates
A 20 mM solution of azidoHiBiT was prepared in water. An aliquot of this solution (2 equiv) was added to dibenzocyclooctyne‐labeled dmQBI‐139 and QBI‐139 (1 equiv) in DPBS without Ca2+/Mg2+. The reaction mixture was incubated overnight with slight agitation. Unreacted peptide was separated from the dmQBI‐139–HiBiT and QBI‐139–HiBiT products using a Micro Bio‐Spin P‐6 column from Bio‐Rad (Waltham, MA) (product #732‐6221) and exchanged into DPBS without Ca2+/Mg2+.
Cellular Uptake and Stability of HiBiT Conjugates
HEK 293 AAV cells, which constitutively produce cytosolic LgBiT, were seeded at 10,000 cells per well in a cell‐culture, flat clear‐bottom, 96‐well white plate (Fisher Scientific, product 07‐000‐167) 24 h before treatment. Cells were then treated with either dmQBI‐139–HiBiT, QBI‐139–HiBiT, or a HiBiT peptide. After 24 h, the medium was removed, and the cells were washed (2×) with DPBS without Ca2+/Mg2+. Then, 100 μL of FluoroBrite DMEM (Thermo Fisher Scientific, product #A1896701) containing FBS (10% v/v) and penicillin–streptomycin solution (1% v/v) was added to each well. Luminescence detection was performed using a Nano‐Glo live cell assay kit according to the manufacturer's instructions (Promega, Madison, WI, USA). Luminescence was measured with a Tecan Spark multimode microplate reader. RLU was quantified at 30 min to represent uptake of the variants at 24 h across two concentrations. The signal from cells plus Furimazine substrate was subtracted as background from the dmQBI‐139–HiBiT, QBI‐139–HiBiT, and HiBiT peptide conditions.
HEK 293 AAV cells constitutively producing cytosolic LgBiT were seeded at 10,000 cells per well in a cell‐culture flat clear‐bottom 96‐well white plate (Thermo Fisher Scientific, product #07‐000‐167). After 24 h, cells were treated with 100 nM dmQBI‐139–HiBiT, QBI‐139–HiBiT, or control HiBiT peptide for 14 h. This concentration was the minimum yielding a detectable signal after 12 h in our system. After 14 h, the medium was removed, and cells were washed (2×) with DPBS without Ca2+/Mg2+. Then, 100 μL of FluoroBrite DMEM (Thermo Fisher Scientific product #A1896701) containing FBS (10% v/v) and penicillin–streptomycin solution (1% v/v) was added to each well. Nano‐Glo Endurazine live‐cell substrate (Promega, Madison, WI, USA) was added to the fresh medium according to the manufacturer's instructions. Luminescence was tracked with a Tecan Spark multimode microplate reader pre‐warmed to 37°C. Cells were maintained in a cell incubator at 37°C, humidified with CO2(5% v/v), and transferred to the plate reader for time‐point analysis. The time points were 2, 4, 6, 8, 14, 24, 34, 48, and 58 h after substrate addition, corresponding to 16, 18, 20, 24, 26, 38, 48, 62, and 72 h of incubation with dmQBI‐139–HiBiT, QBI‐139–HiBiT, or HiBiT. The signal from cells plus Endurazine substrate was subtracted from the signals from dmQBI‐139–HiBiT, QBI‐139–HiBiT, and HiBiT treatments. Each time point represents four replicates per condition.
Results and Discussion
Strategy for Lysine Modification
As a reliable strategy for chemical lysine modification, we sought a method that is gentle, rapid, and selective while adding minimal mass to the protein. We turned our attention to reductive methylation, which adds only six atoms to an amino group (Figure1). We prioritized reagents that accommodate mild labeling conditions, unlike sodium cyanoborohydride, which requires harsh reaction conditions and often yields low conversion rates [47]. We used a reductive methylation procedure adapted from protein crystallization methods that, to our knowledge, has not been applied beyond structural analysis [15]. Our reaction conditions tolerated 1–2 mg mL−1of protein, were performed in 50 mM sodium phosphate buffer, pH 7.5, at 4°C, and were completed in <6 h.
Both RNase 1 and QBI‐139 have eight lysine residues and an N terminus; thus, complete labeling should result in a +252 Da shift. In our hands, we observed complete labeling of both RNase 1 and QBI‐139 upon analysis by Q‐TOF mass spectrometry (Figure2). Hereinafter, we will refer to dimethylated RNase 1 and dimethylated QBI‐139 as dmRNase 1 and dmQBI‐139, respectively.
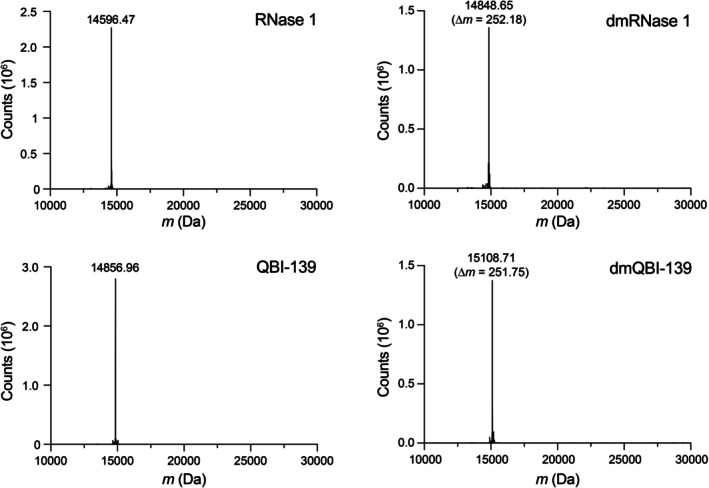
Q‐TOF mass spectra before and after reductive methylation. Each protein has 9 amino groups from 8 lysine residues and an N terminus.N,N‐Dimethylation yields an expected ∆mof 252.49 Da.
Effect of Reductive Methylation on Thermostability
To confirm this strategy as a viable option for biological applications, we first sought to characterize any destabilizing effects on thermostability from the newly introduced methyl groups. We expected that the strategy would not affectTmbecause lysine residues are generally exposed to solvent in folded proteins [48]. Indeed, we observed a shift of ≤1.5°C inTmbetween the unmodified and dimethylated proteins (Figure3).
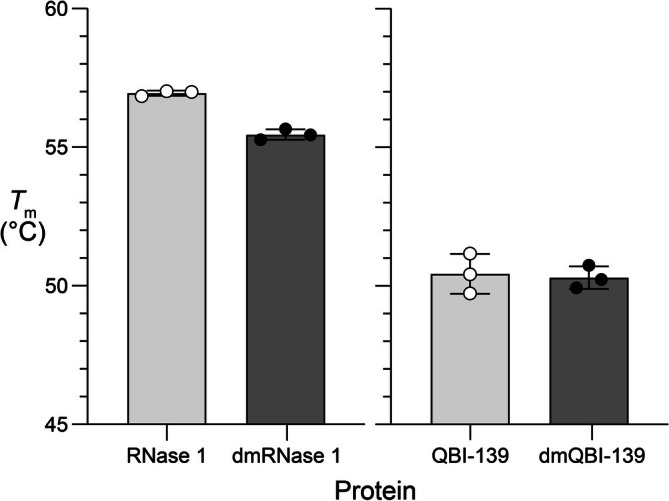
Effect of reductive methylation on thermostability. Values ofTm(mean ± SD) were determined by differential scanning fluorimetry in 100 mM Tris–HCl buffer, pH 7.5, containing NaCl (100 mM). RNase 1, 57.0°C ± 0.1°C; dmRNase 1, 55.5°C ± 0.2°C; QBI‐139, 50.4°C ± 0.7°C; and dmQBI‐139, 50.3°C ± 0.4°C.
Effect of Reductive Methylation on a Protein–Protein Interaction
RNase 1 binds to its cognate inhibitor protein, RI, with femtomolar affinity [49]. In contrast, QBI‐139 has an affinity in the high picomolar/low nanomolar range due to engineered steric and electrostatic interactions [38,50]. For experimental ease, we opted to use QBI‐139 as the model for binding to RI using an assay that exploits a pH‐sensitive fluorescein conjugate whose fluorescence increases upon release from the anionic surface of RI (pI4.7 [51]) into bulk solution [41]. We found that dmQBI‐139 had aKdof 1.8 ± 0.3 nM, compared with 0.64 ± 0.08 nM for unmodified QBI‐139 (Figure4), which is indistinguishable from theKdvalue reported previously for the RI·QBI‐139 complex [37,38]. Thus, adding 18 CH2groups has only a modest effect on a protein–protein interaction. This tolerance likely reflects the inherent flexibility of interfacial lysine residues, which can reorganize to accommodate the additional steric bulk while maintaining their long‐range electrostatic contributions. Conversely, reductive methylation could serve as a chemical probe to identify specific interfaces where a critical lysine residue is deeply buried or strictly constrained, because inserting methyl groups there would disrupt binding.
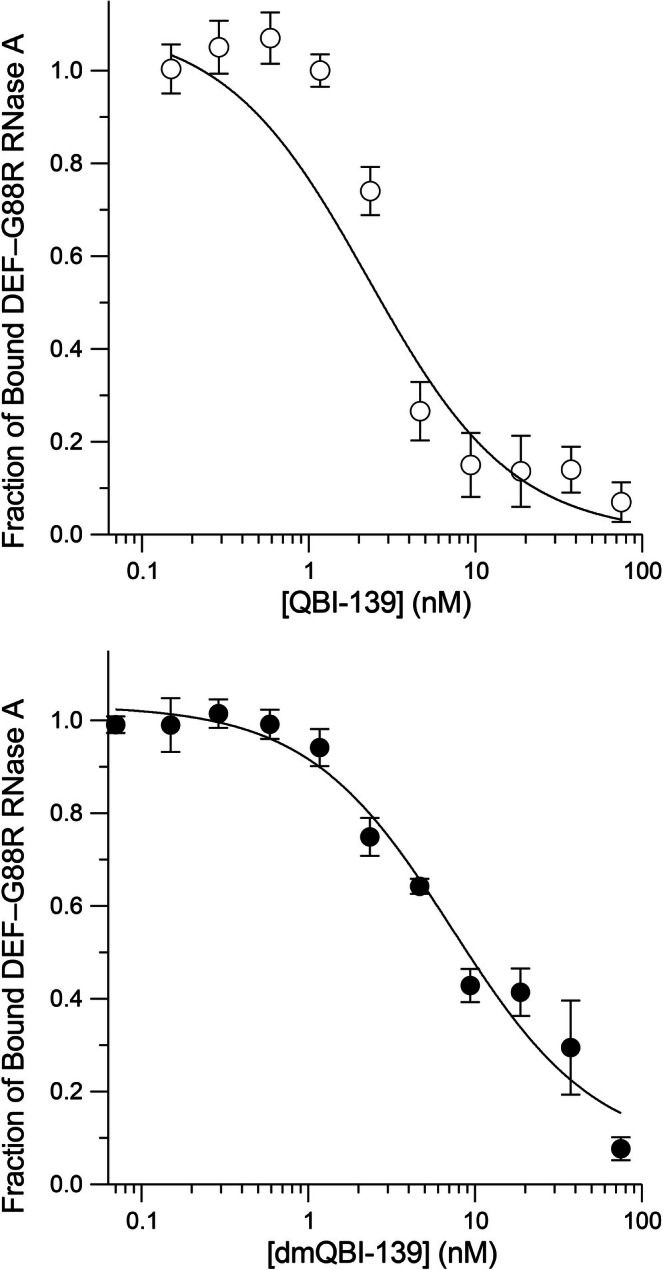
Effect of reductive methylation on a protein–protein interaction. The affinity of QBI‐139 and dmQBI‐139 for RI was measured with a competition assay in PBS containing BSA (0.1 mg mL−1) and DTT (10 mM). Values ofKd(mean ± SD,n= 3) are QBI‐139, 0.64 ± 0.08 nM, and dmQBI‐139, 1.8 ± 0.3 nM.
Effect of Reductive Methylation on Protein Function
RNase 1 relies primarily on three residues to catalyze the cleavage of a P–O5′′bond in RNA: His12, Lys41, and His119 [40]. Among these residues, a hydrogen bond from Lys41 is key to stabilizing the transition state [16]. In addition, RNase 1 relies on favorable interactions between phosphoryl groups and two other conserved lysine residues: Lys7 and Lys66 [17,39,40]. Using 6‐FAM–dArU(dA)2–6‐TAMRA, which is a fluorogenic substrate [52], we determined that RNase 1 and dmRNase 1 havekcat/KMvalues of (3.10 ± 0.19) × 106and (1.62 ± 0.03) × 104M−1s−1, respectively. Thus, RNase 1 retains only 0.14% of its catalytic activity upon dimethylation. For comparison, replacement of Lys41 in RNase 1 with arginine reduced catalytic activity to 1.6% [53]. The effect of dimethylation on catalysis by QBI‐139 is even more severe. Thekcat/KMdecreased from (1.73 ± 0.03) × 105M to (6.0 ± 0.2) × 101M−1s−1, a retention of 0.035%.
QBI‐139 is known to have enhanced cell‐entry abilities and a weaker association with RI than wild‐type RNase 1 [37,38]. To assess cytotoxicity, we opted to use K‐562 cells, as they are highly sensitive to ribonucleases [54]. We found the IC50of QBI‐139 to be 2.3 ± 0.2 μM, whereas dmQBI‐139 was not cytotoxic across the tested concentration range (Figure5). We also assessed toxicity in another leukemia cell line, HAP1, which is nearly haploid and produces fewer copies of proteins than a diploid cell, potentially reducing compensatory effects. In HAP1 cells, we did observe toxicity for dmQBI‐139. The IC50values for the unmodified and dimethylated proteins were 6.5 ± 1.3 and 301 ± 43 μM, respectively. The higher IC50value of dmQBI‐139 is consistent with its diminished ribonucleolytic activity, which is the basis for cytotoxicity [55].
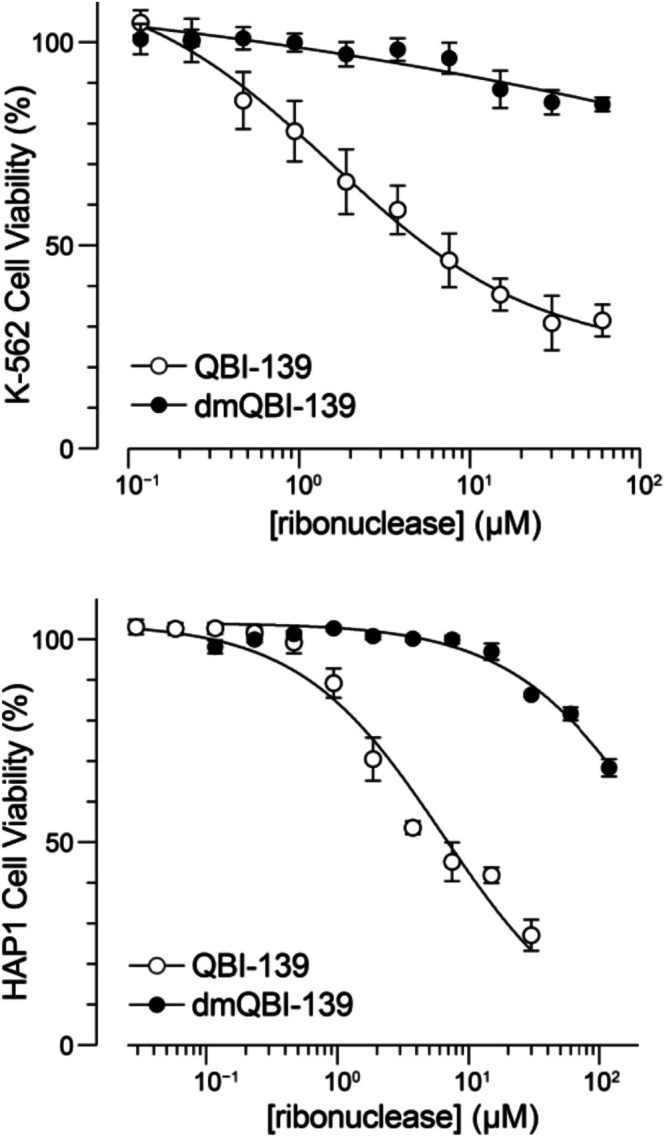
Effect of reductive methylation on the toxicity of ribonucleases for two human cell lines. Values of IC50(mean ± SD,n= 3) are K‐562: QBI‐139, 2.3 ± 0.2 μM; dmQBI‐139, >100 μM; HAP1: QBI‐139, 6.5 ± 1.3 μM; and dmQBI‐139, 301 ± 43 μM.
Effect of Reductive Methylation on Cellular Uptake and Stability
To test for differences in cellular uptake and stability, we implemented a split luciferase assay [56,57]. The HiBiT fragment contains two lysine residues that would be modified by our reductive methylation protocol if fused genetically. Hence, we developed a semisynthetic strategy to conjugate HiBiT to QBI‐139afterreductive methylation. We engineered a variant of QBI‐139 to incorporate a cysteine at residue 19, a modification that does not affect catalysis or RI‐affinity [41,50,58,59]. After reductive methylation, weS‐alkylated Cys19 with DBCO‐PEG4‐maleimide and conjugated the DBCO moiety to azido‐HiBiT via strain‐promoted azide–alkyne cycloaddition. The resulting dmQBI‐139–HiBiT conjugate and its unmodified analog were then used to treat human embryonic kidney (HEK) 293 AAV cells that constitutively produce cytosolic LgBiT [60]. We observed no significant difference in uptake at 24 h (Figure6A); however, during the half‐life tracking studies, we observed a slight initial increase in uptake for dmQBI‐139–HiBiT, perhaps due to its greater hydrophobicity (Figure1) [61].
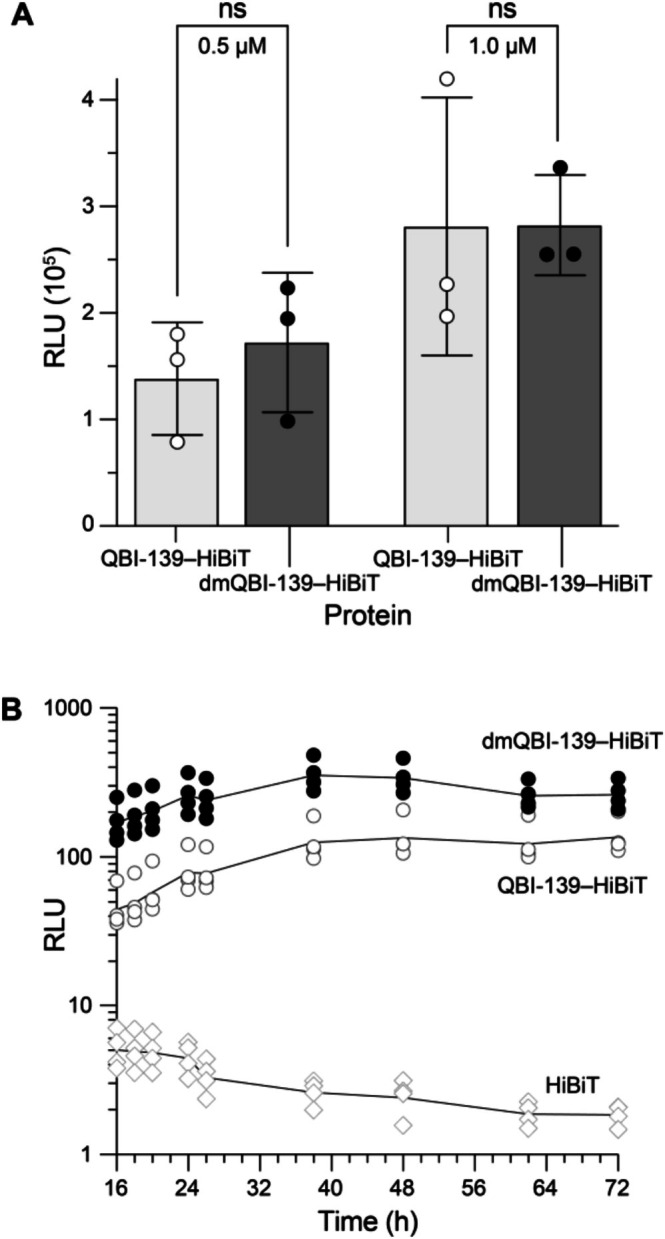
Effect of reductive methylation on cellular uptake and stability using protein–HiBiT conjugates and HEK 293 AAV LgBiT cells. (A) Luminescence observed after 24 h of incubation from two protein concentrations. Values are the mean ± SD (n= 3). (B) Luminescence observed over time after incubation with protein–HiBiT conjugates or HiBiT peptide (100 nM). The lines trace the mean value (n= 4).
Both unmodified and dimethylated QBI‐139–HiBiT persisted in cells with little decay over 72 h (Figure6B). This persistence is likely attributable to the high intrinsic stability of ribonucleases, given that our control HiBiT treatment showed a detectable loss of signal over time. The higher level of dmQBI‐139–HiBiT compared with QBI‐139–HiBiT persisted throughout the time course.
The orthogonality of an engineered cysteine within a dimethylated scaffold presents an opportunity for further protein and peptide engineering. Following the global reductive methylation of all native amino groups, a unique cysteine residue could be modified with a reagent such as 2‐bromoethylamine. This “cysteine elaboration” yieldsS‐(2‐aminoethyl)cysteine (γ‐thialysine), installing a lysine isostere at a user‐defined position [16,62]. Such a strategy would provide an amino group for site‐specific bioconjugation or ubiquitination on an otherwise fully protected scaffold.
Conclusions
Reductive methylation converts every primary amino group in a protein to a tertiary dimethylamino group under mild aqueous conditions within 6 h, adding only 6 atoms per site without affecting other functionality. Using RNase 1 and a cytotoxic variant, QBI‐139, we characterized this modification as a physicochemically conservative alternative to the standard genetic strategy of replacing lysine residues with arginine [15].
Dimethylation preserved thermostability (ΔTm≤ 1.5°C) and had only a modest effect on a protein–protein interaction, increasing theKdfor the RI complex by 3‐fold. Catalytic activity, in contrast, was diminished by 102‐ to 103‐fold, consistent with the steric sensitivity of the ribonuclease active site. This pattern—preservation of global properties, disruption of active‐site function—is informative: it suggests that reductive methylation will be well tolerated in applications that depend on protein stability and binding, such as bioPROTACs, in which efficacy depends on target recruitment rather than enzymatic catalysis. The modification was fully compatible with downstream thiol–maleimide and strain‐promoted azide–alkyne cycloaddition chemistry, supporting its integration into multistep bioconjugation workflows.
Unlike site‐directed mutagenesis, reductive methylation also modifies the N‐terminalα‐amino group, which is itself a substrate for ubiquitination [11,30]. A lysine‐free protein with an acetylated N terminus has been shown to have an increased half‐life [13], but genetic approaches alone cannot modify this site. Reductive methylation addresses that limitation.
Notably, both unmodified and dimethylated ribonucleases showed no measurable decay in signal over 72 h in a live‐cell luminescence assay, indicating that the intrinsically stable ribonuclease fold resists intracellular degradation regardless of methylation status. A protein with lower intrinsic stability would likely be required to resolve kinetic differences between unmodified and dimethylated lysine residues and could be the subject of further study. As tools for intracellular protein delivery continue to advance, reductive methylation offers a practical, generalizable chemical strategy to enhance the intracellular performance of protein‐based therapeutics, including bioPROTACs.
Funding
C.S.G. was supported by a National Defense Science and Engineering Graduate Fellowship sponsored by the US Air Force Research Laboratory. E.C.W. was supported by a Graduate Research Fellowship from the US National Science Foundation. This work was supported by the US National Institutes of Health (R35 GM148220 and P30 CA014051).
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
We are grateful to Dr. Laura E. Strong (Quintessence Biosciences) for QBI‐139, Prof. George M. Church (Harvard Medical School) for HEK 293AAV LgBiT cells, Dr. Luke D. Lavis (University of Wisconsin–Madison) for DEFIA, and the Robert A. Swanson (1969) Biotechnology Center of the Koch Institute for Integrative Cancer Research at MIT for K‐562 and HAP1 cells. We are thankful to Gabriel Saenz (MIT) for help with analytical HPLC.
Molina O. J., Gutierrez C. S., Yang J., Wralstad E. C., and Raines R. T., “Reductive Methylation: An Alternative to Lysine → Arginine Mutagenesis,” Journal of Peptide Science 32, no. 7 (2026): e70110, 10.1002/psc.70110.
Data Availability Statement
The data that support the findings of this study are available in theSupporting Informationof this article.
Associated Data
Data Availability Statement
The data that support the findings of this study are available in theSupporting Informationof this article.
References
- Walsh C. T., Garneau‐Tsodikova S., and G. J. Gatto, Jr. , “Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications,” Angewandte Chemie International Edition 44 (2005): 7342–7372. doi.org/10.1002/anie.200501023
- Walsh G. and Jefferis R., “Post‐Translational Modifications in the Context of Therapeutic Proteins,” Nature Biotechnology 24 (2006): 1241–1252. doi.org/10.1038/nbt1252
- Ryšlavá H., Doubnerová V., Kavan D., and Vaněk O., “Effect of Posttranslational Modifications on Enzyme Function and Assembly,” Journal of Proteomics 92 (2013): 80–109. doi.org/10.1016/j.jprot.2013.03.025
- Keenan E. K., Zachman D. K., and Hirschey M. D., “Discovering the Landscape of Protein Modifications,” Molecular Cell 81 (2021): 1868–1878. doi.org/10.1016/j.molcel.2021.03.015
- Wheadon S. and Cole P. A., “Protein Posttranslational Modifications,” in Advanced Chemical Biology: Chemical Dissection and Reprogramming of Biological Systems, eds. Hang H. C., Pratt M. R., and Prescher J. A. (Wiley–VCH, 2023).
- Clarke S. G., “Protein Methylation at the Surface and Buried Deep: Thinking Outside the Histone Box,” Trends in Biochemical Sciences 38 (2013): 94–102. doi.org/10.1016/j.tibs.2013.02.004
- Biggar S. R. and Li S. S., “Non‐Histone Protein Methylation as a Regulator of Cellular Signalling and Function,” Nature Reviews. Molecular Cell Biology 16 (2015): 5–17. doi.org/10.1038/nrm3915
- Luo M., “Chemical and Biochemical Perspectives of Protein Lysine Methylation,” Chemical Reviews 118 (2018): 6656–6705. doi.org/10.1021/acs.chemrev.8b00008
- Wang Z. A. and Cole P. A., “The Chemical Biology of Reversible Lysine Post‐Translational Modifications,” Cell Chemical Biology 27 (2020): 953–969. doi.org/10.1016/j.chembiol.2020.07.002
- Kitamura N. and Galligan J. J., “A Global View of the Human Post‐Translational Modification Landscape,” Biochemical Journal 480 (2023): 1241–1265. doi.org/10.1042/BCJ20220251
- Breitschopf K., Bengal E., Ziv T., Admon A., and Ciechanover A., “A Novel Site for Ubiquitination: The N‐Terminal Residue, and Not Internal Lysines of MyoD, Is Essential for Conjugation and Degradation of the Protein,” EMBO Journal 17 (1998): 5964–5973. doi.org/10.1093/emboj/17.20.5964
- Thrower J. S., Hoffman L., Rechsteiner M., and Pickart C. M., “Recognition of the Polyubiquitin Proteolytic Signal,” EMBO Journal 19 (2000): 94–102. doi.org/10.1093/emboj/19.1.94
- van de Kooij B., de Vries E., Rooswinkel R. W., et al., “N‐Terminal Acetylation Can Stabilize Proteins Independent of Their Ubiquitination,” Scientific Reports 13 (2023): 5333. doi.org/10.1038/s41598-023-32380-3
- Rayment I., “Reductive Alkylation of Lysine Residues to Alter Crystallization Properties of Proteins,” Methods in Enzymology 276 (1997): 171–179.
- Walter T. S., Meier C., Assenberg R., et al., “Lysine Methylation as a Routine Rescue Strategy for Protein Crystallization,” Structure 14 (2006): 1617–1622. doi.org/10.1016/j.str.2006.09.005
- Messmore J. M., Fuchs D. N., and Raines R. T., “Ribonuclease A: Revealing Structure–Function Relationships With Semisynthesis,” Journal of the American Chemical Society 117 (1995): 8057–8060. doi.org/10.1021/ja00136a001
- Fisher B. M., Ha J. H., and Raines R. T., “Coulombic Forces in Protein–RNA Interactions: Binding and Cleavage by Ribonuclease A and Variants at Lys7, Arg10, and Lys66,” Biochemistry 37 (1998): 12121–12132. doi.org/10.1021/bi980743l
- Caussinus E., Kanca O., and Affolter M., “Fluorescent Fusion Protein Knockout Mediated by Anti‐GFP Nanobody,” Nature Structural & Molecular Biology 19 (2011): 117–121. doi.org/10.1038/nsmb.2180
- Portnoff A. D., Stephens E. A., Varner J. D., and DeLisa M. P., “Ubiquibodies, Synthetic E3 Ubiquitin Ligases Endowed With Unnatural Substrate Specificity for Targeted Protein Silencing,” Journal of Biological Chemistry 289 (2014): 7844–7855. doi.org/10.1074/jbc.M113.544825
- Ludwicki M. B., Li J., Stephens E. A., et al., “Broad‐Spectrum Proteome Editing With an Engineered Bacterial Ubiquitin Ligase Mimic,” ACS Central Science 5 (2019): 852–866. doi.org/10.1021/acscentsci.9b00127
- Simpson L. M., Macartney T. J., Nardin A., et al., “Inducible Degradation of Target Proteins Through a Tractable Affinity‐Directed Protein Missile System,” Cell Chemical Biology 27 (2020): 1164–1180. doi.org/10.1016/j.chembiol.2020.06.013
- Lim S., Khoo R., Peh K. M., et al., “bioPROTACs as Versatile Modulators of Intracellular Therapeutic Targets Including Proliferating Cell Nuclear Antigen (PCNA),” Proceedings. National Academy of Sciences. United States of America 117 (2020): 5791–5800. doi.org/10.1073/pnas.1920251117
- VanDyke D., Taylor J. D., Kaeo K. J., Hunt J., and Spangler J. B., “Biologics‐Based Degraders—An Expanding Toolkit for Targeted‐Protein Degradation,” Current Opinion in Biotechnology 78 (2022): 102807. doi.org/10.1016/j.copbio.2022.102807
- Vukovic D., Winkelvoss D., Udovcic A., et al., “Quantitative Degradation Rate Assessment of bioPROTACs Based on Peptide Degrons, E3 Domains, Adapters and Conjugated Small Molecules,” ACS Chemical Biology 21 (2026): 62–73. doi.org/10.1021/acschembio.5c00569
- Grantham R., “Amino Acid Difference Formula to Help Explain Protein Evolution,” Science 185 (1974): 862–864. doi.org/10.1126/science.185.4154.862
- Bordo D. and Argos P., “Suggestions for “Safe” Residue Substitutions in Site‐Directed Mutagenesis,” Journal of Molecular Biology 217 (1991): 721–729. doi.org/10.1016/0022-2836(91)90528-e
- Henikoff S. and Henikoff J. G., “Amino Acid Substitution Matrices From Protein Blocks,” Proceedings. National Academy of Sciences. United States of America 89 (1992): 10915–10919. doi.org/10.1073/pnas.89.22.10915
- H. K. Hall, Jr. , “Correlation of Base Strengths of Amines,” Journal of the American Chemical Society 79 (1957): 5441–5444.
- Angyal S. J. and Warburton W. K., “The Basic Strengths of Methylated Guanidines,” Journal of the Chemical Society (1951): 2492–2494.
- Ben‐Saadon R., Fajerman I., Ziv T., Hellman U., Schwartz A. L., and Ciechanover A., “The Tumor Suppressor Protein p16(INK4a) and the Human Papillomavirus Oncoprotein‐58 E7 Are Naturally Occurring Lysine‐Less Proteins That Are Degraded by the Ubiquitin System. Direct Evidence for Ubiquitination at the N‐Terminal Residue,” Journal of Biological Chemistry 279 (2004): 41414–41421. doi.org/10.1074/jbc.M407201200
- Hershko A., Heller H., Elias S., and Ciechanover A., “Components of Ubiquitin–Protein Ligase System. Resolution, Affinity Purification, and Role in Protein Breakdown,” Journal of Biological Chemistry 258 (1983): 8206–8214.
- Chau V., Tobias J. W., Bachmair A., et al., “A Multiubiquitin Chain Is Confined to Specific Lysine in a Targeted Short‐Lived Protein,” Science 243 (1989): 1576–1583. doi.org/10.1126/science.2538923
- Haas A. L., Rezelman A., Wefes I., and Pickart C. M., “CytochromecUbiquitination by E2‐14k,” Journal of Biological Chemistry 265 (1990): 23267–23275.
- Lu Y., Lee B.‐H., King R. W., Finley D., and Kirschner M. W., “Substrate Degradation by the Proteasome: A Single‐Molecule Kinetic Analysis,” Science 348 (2015): 200. doi.org/10.1126/science.1250834
- Strong L. E., Kink J. A., Pensinger D., Mei B., Shahan M., and Raines R. T., “Efficacy of Ribonuclease QBI‐139 in Combination With Standard of Care Therapies,” Cancer Research 72, no. Suppl. 1 (2012): 1838.
- Strong L. E., Kink J. A., Mei B., Shahan M. N., and Raines R. T., “First in Human Phase I Clinical Trial of QBI‐139, a Human Ribonuclease Variant, in Solid Tumors,” Journal of Clinical Oncology 30, no. Suppl. 1 (2012): TPS3113.
- Hoang T. T., Tanrikulu I. C., Vatland Q. A., Hoang T. M., and Raines R. T., “A Human Ribonuclease Variant and ERK‐Pathway Inhibitors Exhibit Highly Synergistic Toxicity for Cancer Cells,” Molecular Cancer Therapeutics 17 (2018): 2622–2632. doi.org/10.1158/1535-7163.MCT-18-0724
- Windsor I. W., Dudley D. M., O'Connor D. H., and Raines R. T., “Ribonuclease Zymogen Induces Cytotoxicity Upon HIV‐1 Infection,” AIDS Research and Therapy 18 (2021): 77. doi.org/10.1186/s12981-021-00399-z
- Nogues M. V., Moussaoui M., Boix E., Vilanova M., Ribo M., and Cuchillo C. M., “The Contribution of Noncatalytic Phosphate‐Binding Subsites to the Mechanism of Bovine Pancreatic Ribonuclease A,” Cellular and Molecular Life Sciences 54 (1998): 766–774. doi.org/10.1007/s000180050205
- Raines R. T., “Ribonuclease A,” Chemical Reviews 98 (1998): 1045–1066. doi.org/10.1021/cr960427h
- Lavis L. D., Rutkoski T. J., and Raines R. T., “Tuning the pKaof Fluorescein to Optimize Binding Assays,” Analytical Chemistry 79 (2007): 6775–6782. doi.org/10.1021/ac070907g
- Ressler V. T., Mix K. A., and Raines R. T., “Esterification Delivers a Functional Enzyme Into a Human Cell,” ACS Chemical Biology 14 (2019): 599–602. doi.org/10.1021/acschembio.9b00033
- Wralstad E. C., Sayers J., Fittolani G., et al., “Sacrificial Methionine Residues Protect Active‐Site Histidines From Oxidation,” Submitted, (2026).
- Lomax J. E., Bianchetti C. M., Chang A., G. N. Phillips, Jr. , Fox B. G., and Raines R. T., “Functional Evolution of Ribonuclease Inhibitor: Insights From Birds and Reptiles,” Journal of Molecular Biology 426 (2014): 3041–3056. doi.org/10.1016/j.jmb.2014.06.007
- Wralstad E. C. and Raines R. T., “Sensitive Detection of SARS‐CoV‐2 Main Protease 3CLpro With an Engineered Ribonuclease Zymogen,” Protein Science 33 (2024): e4916. doi.org/10.1002/pro.4916
- Cory A. H., Owen T. C., Barltrop J. A., and Cory J. G., “Use of an Aqueous Soluble Tetrazolium/Formazan Assay for Cell Growth Assays in Culture,” Cancer Communications 3 (1991): 207–212. doi.org/10.3727/095535491820873191
- Jentoft N. and Dearborn D. G., “Labeling of Proteins by Reductive Methylation Using Sodium Cyanoborohydride,” Journal of Biological Chemistry 254 (1979): 4359–4365.
- Savojardo C., Manfredi M., Martelli P. L., and Casadio R., “Solvent Accessibility of Residues Undergoing Pathogenic Variations in Humans: From Protein Structures to Protein Sequences,” Frontiers in Molecular Biosciences 7 (2020): 626363. doi.org/10.3389/fmolb.2020.626363
- Dickson K. A., Haigis M. C., and Raines R. T., “Ribonuclease Inhibitor: Structure and Function,” in Progress in Nucleic Acid Research and Molecular Biology, vol. 80 (Elsevier, 2005), 349–374. doi.org/10.1016/S0079-6603(05)80009-1
- Johnson R. J., Lavis L. D., and Raines R. T., “Intraspecies Regulation of Ribonucleolytic Activity,” Biochemistry 46 (2007): 13131–13140. doi.org/10.1021/bi701521q
- Blackburn P., Wilson G., and Moore S., “Ribonuclease Inhibitor From Human Placenta. Purification and Properties,” Journal of Biological Chemistry 252 (1977): 5904–5910.
- Kelemen B. R., Klink T. A., Behlke M. A., Eubanks S. R., Leland P. A., and Raines R. T., “Hypersensitive Substrate for Ribonucleases,” Nucleic Acids Research 27 (1999): 3696–3701. doi.org/10.1093/nar/27.18.3696
- Leland P. A., Staniszewski K. E., Kim B.‐M., and Raines R. T., “Endowing Human Pancreatic Ribonuclease With Toxicity for Cancer Cells,” Journal of Biological Chemistry 276 (2001): 43095–43102. doi.org/10.1074/jbc.M106636200
- Haigis M. C., Kurten E. L., and Raines R. T., “Ribonuclease Inhibitor as an Intracellular Sentry,” Nucleic Acids Research 31 (2003): 1024–1032. doi.org/10.1093/nar/gkg163
- Rutkoski T. J., Kurten E. L., Mitchell J. C., and Raines R. T., “Disruption of Shape‐Complementarity Markers to Create Cytotoxic Variants of Ribonuclease A,” Journal of Molecular Biology 354 (2005): 41–54. doi.org/10.1016/j.jmb.2005.08.007
- Okano S., Kawaguchi Y., Kawano K., Hirose H., Imanishi M., and Futaki S., “Split Luciferase‐Based Estimation of Cytosolic Cargo Concentration Delivered Intracellularly via Attenuated Cationic Amphiphilic Lytic Peptides,” Bioorganic & Medicinal Chemistry Letters 72 (2022): 128875. doi.org/10.1016/j.bmcl.2022.128875
- Okon A., Yang J., Giancola J. B., et al., “Facile Access to Branched Multispecific Proteins,” Bioconjugate Chemistry 35 (2024): 954–962. doi.org/10.1021/acs.bioconjchem.4c00162
- Johnson R. J., Chao T. Y., Lavis L. D., and Raines R. T., “Cytotoxic Ribonucleases: The Dichotomy of Coulombic Forces,” Biochemistry 46 (2007): 10308–10316. doi.org/10.1021/bi700857u
- Lomax J. E., Eller C. H., and Raines R. T., “Comparative Functional Analysis of Ribonuclease 1 Homologs: Molecular Insights Into Evolving Vertebrate Physiology,” Biochemical Journal 474 (2017): 2219–2233. doi.org/10.1042/BCJ20170173
- Chao G., Travis C., and Church G., “Measurement of Large Serine Integrase Enzymatic Characteristics in HEK293 Cells Reveals Variability and Influence on Downstream Reporter Expression,” FEBS Journal 288 (2021): 6410–6427. doi.org/10.1111/febs.16037
- Calabretta L. O., Yang J., and Raines R. T., “Nα‐Methylation of Arginine: Implications for Cell‐Penetrating Peptides,” Journal of Peptide Science 29 (2023): e3468. doi.org/10.1002/psc.3468
- Raftery M. A. and Cole R. D., “On the Aminoethylation of Proteins,” Journal of Biological Chemistry 241 (1966): 3457–3461.
Republished from the open web under CC-BY. Authors: Molina OJ, Gutierrez CS, Yang J, Wralstad EC, Raines RT. Read the original.