The expanding role of protease therapeutics (2012-2026): from replacement therapies to immune system modulation and beyond.
Proteases are powerful therapeutic agents, offering unique advantages over conventional small molecules and biologics through their ability to directly and precisely cleave and (de)activate their protein targets. Since the early 1990s, FDA-approved protease therapeutics have served as replacement therapies for hematological disorders, as enzyme supplements for digestive disorders, and as treatments for neuromuscular disorders. Recent developments have expanded the use of native proteases to modulate antibody responses, improve transplant outcomes, and treat rare conditions with high unmet needs. Despite challenges such as immunogenicity and substrate specificity, the therapeutic landscape of proteases is being redefined by innovations in enzyme engineering and discovery. These advances, combined with targeted delivery strategies and improved stability, are reshaping proteases into precise and adaptable therapeutic agents. Rather than being limited to traditional uses, proteases are increasingly recognized for their potential to address complex conditions such as viral infections, neurodegeneration, and fibrosis, among others. With continued development, proteases are positioned to become a versatile and robust class of biologics with expanding clinical relevance. The present review explores the evolving landscape of protease therapeutics, focusing on their clinical applications, immune-modulatory capabilities, and future potential in precision medicine. This review provides a timely update to the comprehensive article "Proteases as Therapeutics" by Craik, Page, and Madison, published in the Biochemical Journal in 2011.
Introduction
The ability to harness proteases as precise therapeutic agents has long been a compelling goal in medicine. Early perspectives, including a prominent 2011 review by Craik and colleagues, described proteases as a distinct therapeutic class with diverse clinical applications [1,2]. Since then, a growing number of proteolytic enzymes have been approved for clinical use, primarily as enzyme replacement therapies or as agents that act on well-defined extracellular substrates (Table 1). These include treatments for hematological and metabolic disorders as well as drugs that degrade extracellular protein targets, such as ADZYNMA® for congenital thrombotic thrombocytopenic purpura and Voraxaze® for reducing toxic plasma methotrexate levels. Thus far, FDA-approved protease-based therapies have predominantly exploited only a subset of proteases' native biological capabilities.
Table: FDA-approved protease drugs from 2011 to 2025
A central objective in next-generation protease therapeutics is therefore to move beyond enzyme replacement and instead draw inspiration from the regulatory roles proteases playin vivo. Proteases regulate a myriad of cellular processes, from blood coagulation and protein activation/degradation to host defense and adaptive immunity [3,4]. Importantly, they achieve this regulation not by completely degrading proteins, i.e., proteasome enzymes, but by cleaving specific sequences within their targets. Consequently, proteases offer a unique opportunity to selectively eliminate disease-driving proteins, rewire dysregulated biological pathways, or augment innate host defense mechanisms [5–7]. Moreover, compared with antibodies, which are highly effective for systemically accessible targets but act in a strictly stoichiometric manner, enzymes offer a fundamentally different pharmacologic paradigm. As enzymes, proteases can turn over multiple substrate molecules, enabling sub-stoichiometric activity that is particularly well-suited for high-abundance targets or sites that are poorly accessible from circulation [8]. Furthermore, emerging approaches such as mRNA-based delivery of proteases aim to enable transientin vivoproduction of enzymes like ADAMTS13, potentially reducing the need for repeated protein infusions, although these strategies remain in preclinical development [9].
Despite this considerable therapeutic potential, successful clinical translation of any protein-based therapeutic requires overcoming a broad and interconnected set of engineering and pharmacologic challenges. For proteases, these considerations include catalytic performance (kcat/KM), substrate specificity, immunogenicity, stability (resistance to proteolysis), delivery constraints, and manufacturability (Figure 1) [2,8,10]. In parallel, pharmacokinetic properties, such as clearance rate and bioavailability, are critical determinants of dosing feasibility. While many protease therapeutics are administered intravenously to ensure systemic exposure, alternative routes including oral delivery (e.g., proteases targeting dietary antigens such as gluten), topical application, and localized injection have also demonstrated clinical potential [2,11]. Manufacturing considerations likewise play a critical role, as recombinant proteases produced in bacterial or eukaryotic systems must meet stringent purity standards, including rigorous removal of host cell proteins and other contaminants [12]. These requirements contribute significantly to production costs and can influence platform selection during early development. Beyond manufacturing, optimization of protease therapeutics often requires balancing improvements in pharmacokinetic and biophysical properties against preservation of catalytic function. For example, strategies to improve serum half-life, such as PEGylation or Fc fusion, can reduce catalytic efficiency or substrate accessibility, highlighting a fundamental modification–activity trade-off [13–15]. Similarly, achieving high specificity often comes at the expense of catalytic rate or stability, necessitating multi-parameter optimization during development [16,17]. Collectively, these constraints underscore the need for integrated engineering approaches that simultaneously address activity, specificity, stability, and delivery [16,18].
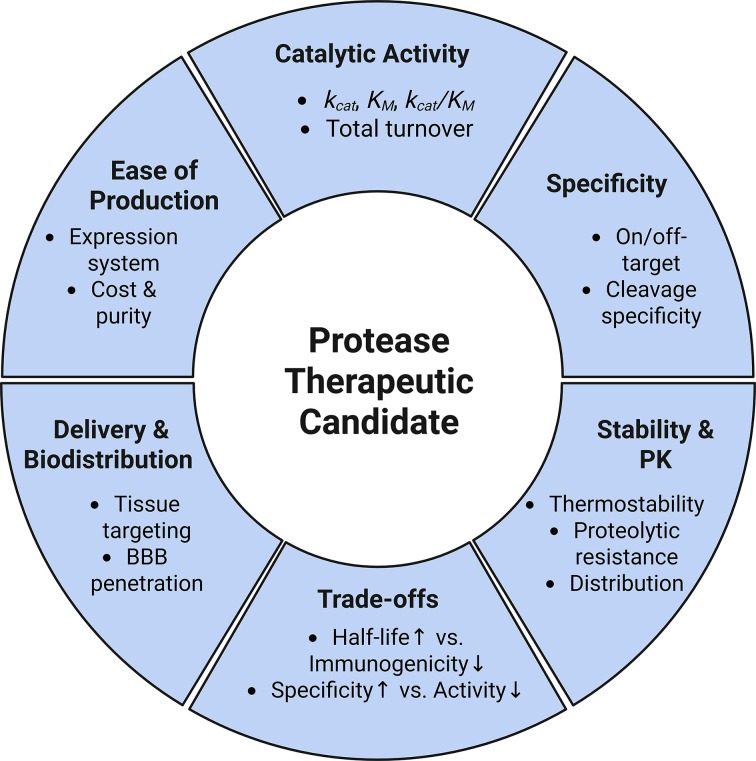
Overview of the factors governing the clinical translation of protease therapeutics including: catalytic activity, specificity, stability/pharmacokinetics, trade-offs, delivery/biodistribution, and ease of production
Within this multidimensional framework, substrate specificity emerges as a particularly important determinant of clinical tractability. A key throughline in the early expansion of the protease pharmacopeia is its reliance on the limited subset of proteases that possess inherently narrow substrate specificities [19,20]. To fully harness the therapeutic potential of proteases, there is a pressing need to overcome the longstanding challenges associated with engineering protease specificity. Recent advances in experimental and computational protein engineering, including AI-driven approaches forde novoprotease design, are rapidly expanding the space of proteases with tailored activity and specificity. These advances are widely viewed as a major driver of innovation in the field [21]. Diffusion-based generative models and related frameworks have begun to overcome long-standing limitations in scaffold selection and specificity engineering, accelerating protease discovery and optimization [22]. These methodological advances, together with recent progress in protease specificity engineering, have been comprehensively reviewed elsewhere [18]. Accordingly, rather than focusing on protease engineering platforms, the present review centers on how proteases are being deployed in recent clinical, preclinical, and emerging therapeutic applications.
As the present review highlights, recent progress in protease therapeutics has been driven less by a single technological advance than by convergent innovation across multiple application domains. In particular, three emerging areas are reshaping the landscape of protease-based medicines: (1) bacterial proteases that cleave immunoglobulins to modulate immune responses and treat autoimmune disease; (2) microbial and plant proteases that selectively degrade pathogenic proteins, including dietary antigens such as gluten and disease-associated substrates implicated in neurodegeneration; and (3) engineered proteases designed to destabilize misfolded or aggregated proteins. Together, these examples illustrate how advances in targeting, specificity, and delivery are enabling proteases to move beyond replacement therapies toward more precise and programmable therapeutic modalities.
Advancing hematological therapies through engineered proteases
Protease-based therapeutics have a long history in the treatment of hematological disorders. Early clinical successes include the replacement of essential coagulation proteases such as factor IX (FIX). Recombinant FIX, marketed as BeneFIX®, was approved by the FDA in 1997 and is effective at treating acute bleeding episodes [2]. Despite its success, FIX replacement therapy requires repeated dosing due to a relatively short circulating half-life (∼18 h), which increases the risk of developing anti-FIX antibodies.
One strategy to address these limitations is to extend protease residence time in circulation by engineering stabilizing molecular interactions. In the coagulation system, fusion of factor VIII (FVIII) to a high-affinity anti–von Willebrand factor (VWF) nanobody (Nb) enhanced FVIII–VWF complex stability by slowing dissociation kinetics, resulting in prolonged circulatory residence time while preserving normal thrombin-mediated proteolytic activation [23].In vivo, this increased complex stability translated into improved hemostatic efficacy and a substantial reduction in anti-FVIII antibody formation. Conceptually, this approach parallels other circulation-time extension strategies, such as albumin binding, but uniquely leverages a native physiological interaction to improve both pharmacokinetics and immunogenicity.
In the past decade, the hematology therapeutic landscape has broadened substantially with the introduction of antibody-based agents and factor-mimetic biologics, including bispecific antibodies that can functionally replace missing coagulation factors [24–26]. These modalities have delivered transformative clinical benefits and now constitute a major focus of therapeutic development. Importantly, however, their emergence has not marked a departure from protease-based strategies. Rather, ongoing efforts to engineer coagulation proteases, such as improved variants of activated recombinant factor VII, reflect continued innovation within the field.
Marzeptacog alfa (activated) (MarzAA) is a variant of activated recombinant human FVII developed for the treatment of episodic bleeding in patients with inherited bleeding disorders [27]. MarzAA has four site-directed amino acid substitutions. Two catalytic-domain mutations (Q286R and M298Q) enhance platelet binding and tissue factor–independent activation of factor X, increasing procoagulant activity [28]. Two additional substitutions (T128N and P129A) introduce an N-linked glycosylation site to extend the circulating half-life [29]. Across two phase I studies (NCT01439971andNCT04072237) and one phase II trial (NCT03407651), MarzAA demonstrated a favorable safety and pharmacokinetic profile following both intravenous and subcutaneous administration, with no serious adverse events or antidrug antibody formation reported [27,29,30]. In a phase II prophylaxis study in patients with hemophilia A/B with inhibitors, daily subcutaneous MarzAA (30 μg/kg with escalation to 60 μg/kg for breakthrough bleeding) produced a marked and statistically significant reduction in annualized bleeding rate, decreasing from a baseline mean of approximately 19.8 to 1.6, along with a substantial reduction in the proportion of days with bleeding. Despite these results, the subsequent phase III study (NCT04489537) was terminated after Catalyst Biosciences discontinued development due to feasibility constraints, including enrollment challenges, increased competition, and widespread adoption of alternative prophylactic therapies [31]. MarzAA was later acquired by GC Biopharma, and its future approval depends on its success in clinical trials [32].
Beyond MarzAA, Catalyst Biosciences has leveraged its engineering platform to advance additional protease therapeutics, including dalcinonacog alfa for subcutaneous treatment of rare bleeding disorders, and CB 4332, an engineered complement factor I (CFI) for systemic prophylaxis in patients with CFI deficiency [33]. However, dalcinonacog alfa was transferred to GC Biopharma as part of the same acquisition as MarzAA in 2023, while catalyst’s complement portfolio, including CB 4332 and related intellectual property, was sold in 2022 to Vertex Pharmaceuticals [32,34].
Despite strong mechanistic rationale and compelling clinical efficacy, the MarzAA program illustrates how advances in protease engineering can be constrained by translational and market forces rather than biological feasibility.
Modulating immune responses through proteolytic deactivation of immunomodulators
Autoimmune diseases encompass a broad spectrum of conditions characterized by dysregulated B-cell and T-cell function [35]. Many of these autoimmune diseases, including systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), are associated with elevated levels of immunoglobulin M (IgM) and immunoglobulin G (IgG) autoantibodies [36]. Current therapeutic strategies focus primarily on broad immunosuppression using agents like corticosteroids, non-steroidal anti-inflammatory drugs, and disease-modifying antirheumatic drugs, but these treatments often come with severe side effects and incomplete disease control [37]. Biologics have made some progress in addressing these challenges. For example, belimumab, a monoclonal antibody targeting B-cell activating factor, can reduce disease activity and organ damage in SLE [38]. However, due to the heterogeneity of IgG-mediated disease, belimumab has limited efficacy, and it is currently not approved for patients with severe active central nervous system lupus [39].
Against this backdrop, proteases are emerging as precision immunomodulators. By selectively degrading immunoglobulins, cytokines, and other immune mediators, proteases can dampen inflammation and limit immune-mediated damage in autoimmune diseases, organ transplantation, and celiac disease. To clarify how proteases can be harnessed in this way, it is useful to consider the shared and divergent features of the major immunoglobulin classes.
Immunoglobulins comprise multiple classes (IgM, IgG, IgA, IgE, and IgD) that share a conserved overall architecture but differ in quaternary structure, localization, and effector function [40]. All antibodies contain two antigen-binding Fab regions and a constant Fc region; however, differences in heavy-chain constant domains give rise to distinct isotypes and subclasses. IgG, the most abundant serum antibody, is further divided into subclasses (IgG1 to IgG4) that vary in hinge length, disulfide bonding, and neonatal Fc receptor (FcRn) interactions. IgA exists primarily as a dimer in mucosal tissues, while IgM forms pentameric structures that enable high-avidity binding. Despite these differences, conserved sequence and structural elements provide accessible cleavage sites that proteases can exploit. For example, IgG-cleaving enzymes such as IdeS recognize conserved motifs within the lower hinge region, enabling broad activity across subclasses, whereas other proteases exhibit selectivity based on subtle sequence or structural differences between isotypes [41]. These shared and divergent features underline how proteases can achieve either pan-immunoglobulin activity or isotype-specific targeting.
Several of these enzymes are now in clinical trials to evaluate their safety and efficacy (Table 2). The following subsections highlight key proteases targeting IgG, IgM, IgA, and immunogenic gluten peptides, outlining their mechanisms, therapeutic promise, and translational challenges.
Table: Protease drugs in development
Harnessing IdeS for immune modulation and transplant success
IdeS (imlifidase), an IgG-degrading cysteine endopeptidase fromStreptococcus pyogenes, cleaves the Fc region of IgG antibodies, blocking FcRn interactions and interfering with immune activation (Figure 2) [42,43]. Repurposed for desensitization in transplantation, IdeS reduces donor-specific antibodies (DSAs) in highly sensitized patients at risk of graft rejection due to pre-existing antibodies against human leukocyte antigens (HLAs) [44]. In one study, 24 of 25 such patients underwent successful HLA-incompatible kidney transplantation with stable renal function maintained post-surgery treatment [43].
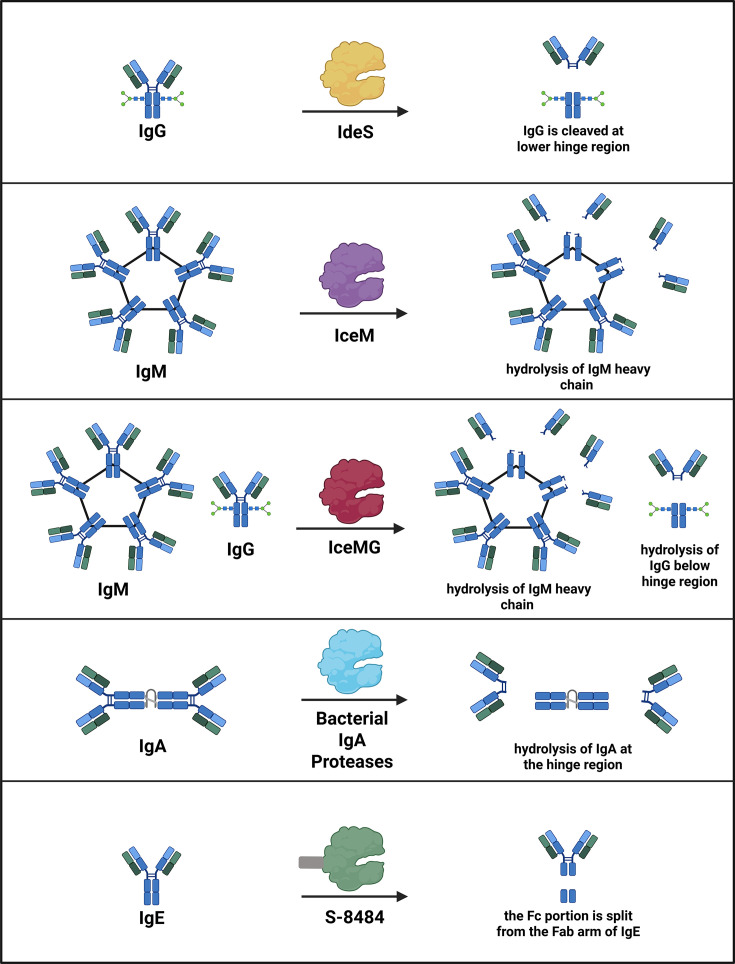
Proteolytic inactivation of immunoglobulins as a strategy to modulate immune responses. Diverse immunoglobulin-targeting proteases cleave IgG, IgM, and IgE at distinct structural sites, generating antibody fragments with characteristic functional outcomes. For example, IgG-degrading enzymes such as IdeS and its engineered derivatives cleave the IgG hinge region, removing Fcγ-mediated effector functions and disrupting FcRn and complement activation. Together, these proteases illustrate how selective cleavage of immunoglobulin isotypes can rapidly and precisely suppress pathogenic humoral immunity across autoimmunity, transplantation, allergy, and gene therapy.
However, since IdeS is derived from a bacterial pathogen, it can trigger the formation of anti-drug antibodies (ADA) and the non-selective cleavage of protective antibodies [45]. Most patients have low-level preexisting anti-IdeS IgG from priorS. pyogenesexposure and develop an IgG response that typically peaks 2 to 3 weeks after treatment [46]. IdeS also has a short half-life (∼5 h), which contributes to a significant limitation: donor-specific HLA antibodies and anti-HLA antibodies often rebound within 3 to 7 days, frequently requiring concurrent immunosuppressive therapy [15,44]. To address IdeS’ short half-life, a monovalent Fc fusion variant (IdeS-Fcmonov) was engineered by genetically fusing IdeS to a heterodimeric human IgG Fc [15]. The Fc fusion extends serum persistence via FcRn-mediated recycling while minimizing Fc effector function [47]. The Fc domain was engineered to be monovalent and inert, preventing unwanted immune activation and protease-mediated self-cleavage. This construct increased circulating half-life approximately seven-fold; however, steric and conformational constraints introduced by the Fc fusion reduced catalytic efficiency relative to wild-type IdeS, highlighting a trade-off between pharmacokinetic enhancement and enzymatic activity [15].
Despite these limitations, IdeS received conditional approval from the European Medicines Agency (EMA) in 2020 to treat kidney transplant recipients with a positive crossmatch [48]. A five-year study of 39 patients showed 90% patient survival and 84% graft survival [49]. More recently, ConfIdeS, a Phase 3 trial conducted by Hansa Biopharma, evaluated 64 highly sensitized kidney transplant patients [50]. Patients randomized to receive IdeS exhibited markedly superior kidney function at 12 months, with a mean estimated glomerular filtration rate of 51.5 ml/min/1.73 m2compared with 19.3 ml/min/1.73 m2in the control arm.
Beyond transplantation, IdeS is in Phase 3 evaluation for anti-glomerular basement membrane (anti-GBM) disease (GOOD-IDES-02 trial) and holds orphan drug designation from both the U.S. FDA and EMA [51]. Hansa Biopharma’s IgG-cleaving enzyme, HNSA-5487, achieves >95% IgG reduction in hours, with a significantly lower ADA response compared with IdeS, suggesting improved potential for redosing [52]. HNSA-5487’s favorable safety profile suggests its potential application in chronic autoimmune diseases, including myelin oligodendrocyte glycoprotein antibody disease, neuromyelitis optica, and myasthenia gravis.
Targeting IgM to overcome immune barriers in gene delivery
Recombinant adeno-associated virus (rAAV) gene therapies face immunological hurdles from IgM and IgG antibodies [53]. rAAV vectors are the leading platform for gene delivery therapy in a variety of diseases, including hemophilia A, hemophilia B, and Crigler–Najjar syndrome [54]. However, neutralizing antibodies against AAVs are present in 30% to 60% of humans, and even low titers can block AAV gene transfer [55]. IgG-cleaving proteases, such as IdeS, IdeZ, and IdeXork, have been explored to overcome anti-AAV immunity and transiently deplete circulating IgG [56,57]. While these enzymes can create a brief window of reduced IgG levels, their efficacy diminishes at higher neutralizing titers, and the rapid development of ADA drastically reduces the effectiveness of redosing attempts [57].
To overcome the IgM-mediated anti-AAV immune response, IceM, an IgM-specific protease fromLachnoanaerobaculum saburreum, was recently identified and optimized [58]. To broaden its utility, a dual-activity enzyme, IceMG, was engineered by fusing IceM to an IgG-cleaving domain. Preclinical studies demonstrate that IceM and IceMG transiently degrade circulating IgM and IgG, effectively mitigating complement activation, a key driver of AAV vector clearance [58]. Notably, IceMG treatment reduced AAV9-induced complement activation within an hour and sustained its effect for up to two weeks, suggesting a potential therapeutic window for preconditioning prior to gene therapy. These proteases may also be useful in the context of autoimmune disease and transplantation. For example, a subset of myasthenia gravis (MG) involves IgM autoantibodies that contribute to disease pathogenesis, meaning IdeS alone would be insufficient, and combination therapy with an IgM-cleaving protease or the engineered IceMG enzyme may be optimal [59]. However, because IgM plays an important role in immune homeostasis by supporting apoptotic cell clearance, limiting excessive inflammation, and regulating autoimmunity, careful safety evaluation will be necessary before advancing these therapeutics toward clinical use [60].
Expanding immunoglobulin targeting with engineered proteases from Seismic Therapeutics
Seismic Therapeutics, a clinical-stage biotechnology company leveraging machine learning-guided design through its IMPACT platform, is advancing a pipeline of protease therapeutics to selectively cleave disease-relevant immunoglobulins. Two lead candidates, S-1117 and S-8484, exemplify this approach by targeting IgG and IgE, respectively [61]. S-1117 is a novel engineered Fc-fused pan-IgG protease designed to address IgG autoantibody-mediated diseases [61]. To improve pharmacokinetics while minimizing unintended immune activation, Seismic Therapeutics linked the protease to a human IgG1 Fc domain that is specifically modified to lack Fc-mediated effector function and resist proteolytic self-cleavage. This design extends serum half-life through FcRn-mediated recycling while maintaining catalytic activity against IgG substrates [59]. Seismic also used their proprietary IMPACT platform to decrease immunogenicity by reducing T and B cell epitopes. Preclinical studies show that S-117 cleaves all IgG subclasses, removes IgG effector functions, and can also cleave membrane-bound IgG on memory B cells [62]. With its reduced immunogenicity and improved half-life, S-1117 may support both acute and chronic dosing, unlike IdeS, which is generally limited to single or infrequent administration. In March 2025, Seismic initiated a randomized, double-blind, placebo-controlled Phase 1 trial (NCT06828393) to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of S-1117 in healthy volunteers.
In parallel, Seismic is advancing S-8484, an engineered Fc-fused IgE protease designed to treat allergic diseases [63]. IgE plays a central role in type I hypersensitivity disorders such as food allergy, asthma, and allergic rhinitis. Furthermore, IgE-mediated food allergy is a major driver of anaphylaxis in children [64]. S-8484 was optimized for selectivity and potency using structure-augmented machine learning, overcoming the promiscuity often associated with natural bacterial proteases. Preclinicalin vitrostudies demonstrated rapid and selective cleavage of soluble IgE, depletion of IgE from B-cell receptors, and down-regulation of high-affinity IgE receptors on mast cells from humanized mice [63].In vivoS-8484 reduced serum IgE and eosinophils in allergen-induced asthma models, attenuated IgE-driven tissue inflammation, and provided rapid resolution of systemic anaphylaxis. These data support ongoing IND-enabling studies and highlight the potential for a protease-based therapeutic to induce durable IgE depletion across multiple allergic indications.
Transforming IgA nephropathy treatment with IgA-degrading proteases
Aberrantly glycosylated immunoglobulin A (agIgA1), a key pathogenic factor in IgA nephropathy (IgAN), arises from aberrant O-glycosylation in the hinge region of IgA1 [65]. These agIgA1s can be recognized by circulating IgG or IgA antibodies, promoting the formation of immune complexes that then trigger inflammation and glomerular damage [66]. Bacterial IgA-cleaving proteases from strains such asNeisseria gonorrhoeae,Haemophilus influenzae, andNeisseria meningitidisspecifically degrade agIgA1 without affecting other proteins like human IgG or bovine serum albumin [65]. Excitingly,in vitroand mouse model studies demonstrate effective degradation of agIgA1 and its immune complexes. Nonetheless, further studies are needed to ensure enzyme specificity, safety, and efficacy in advanced models before clinical translation.
Mechanistic studies of IgAN show that circulating agIgA1 forms complexes with soluble receptor CD89 and anti-agIgA1 autoantibodies. These complexes are retained in the mesangium via interactions with the transferrin receptor, triggering complement activation, inflammation, and progressive glomerular injury [67]. In a humanized mouse model of IgAN, recombinant IgA1-cleaving proteases rapidly reduced circulating IgA1–CD89 complexes and markedly decreased mesangial IgA1 deposition following systemic administration. Repeated dosing further disrupted associated mesangial binding partners, including CD89, transferrin receptor, and transglutaminase 2, leading to reduced complement deposition, inflammatory cell infiltration, fibrosis, and hematuria, thereby demonstrating disease-modifying potential.
Notably, in addition to cleaving IgA1, the meningococcal IgA1-specific serine protease can also degrade human IgG3 [68]. Despite its low abundance in serum (∼7%), IgG3 plays a critical role in antibacterial immune defense, and reduced IgG3 levels are associated with severe bacterial respiratory tract infections [68,69]. These findings underscore the need to carefully evaluate off-target effects when considering IgA1-P as a therapeutic strategy.
Protease supplementation to combat celiac disease
Celiac disease (CeD) is driven by immunogenic gluten peptides resistant to degradation by human digestive enzymes [70]. Bacterial and fungal-derived gluten-degrading enzymes, particularly prolyl endopeptidases (PEPs), show promise in degrading these peptides but face challenges like instability in gastric conditions and incomplete efficacyin vivo[70,71]. Chemical modifications such as PEGylation can enhance stability but may reduce activity or increase immunogenicity [72].
In contrast, a novel endoprotease 40 (E40), derived from the acidophilic actinomyceteActinoallomurusA8, exhibits robust glutenase activity at gastric pH and can resist pepsin degradation, effectively detoxifying gluten peptidesin vitro[73,74]. Proteomic and immunoassay analyses confirmed that E40-treated gastric digestion exhibited a marked reduction in α-gliadin epitopes, known to trigger immune responses in CeD patients. Importantly, residual traces of ω- and γ-gliadin peptides did not elicit a significant interferon (IFN)-γ response in T-cell assays, suggesting effective detoxification of gluten-derived immunogenic sequences. PEPs fromSphingomonas capsulatahave also been optimized using machine learning to improve stability and activity under gastric conditions, with promising mutant variants demonstrating enhanced gluten hydrolysis and potential for oral enzymatic therapy [75].
Repurposing microbial and plant proteases for therapeutic applications
Microbial and plant proteases are attractive therapeutic agents due to their cost-effectiveness, stability, and ease of production compared with animal-derived enzymes [76,77]. Recent advances highlight their potential across diverse medical applications, including neurodegenerative diseases, metabolic disorders, and viral infections. Harnessing their enzymatic capabilities may enable novel treatments for conditions currently lacking effective therapies. Several of these enzymes have already advanced into clinical use or preclinical evaluation (Table 3). The following examples highlight microbial and plant proteases, outlining their proposed mechanisms and therapeutic benefits.
Table: Selected microbial and plant proteases with therapeutic applications
Proteases fromZingiber officinale(ginger) show promise as acetylcholinesterase inhibitors, potentially providing symptomatic relief in Alzheimer’s disease (AD) with superior stability across temperature and pH ranges compared with current drugs [78,79]. Additionally, ginger proteases hydrolyze wheat gluten to produce bioactive peptides that inhibit dipeptidyl peptidase-IV (DPP-IV), a target in type 2 diabetes management [80]. These peptides demonstrate potent inhibitory activity and improve gluten solubility and bioavailability. While some peptide modifications reduce DPP-IV inhibition, they may confer other health benefits, such as hepatoprotection and mood regulation [81].
Microbial proteases, especially extracellular fungal enzymes, demonstrate potential antiviral activity by targeting key viral proteins [82].In silicodocking studies have demonstrated that proteases from the A1A, M20A, S10, S8A, and T1A families can bind to key SARS-CoV-2 viral proteins, such as the spike, envelope, ORF-7a, and Nsp2, with binding energies below −50 kJ/mol [83]. Additionally, clinically approved microbial proteases, likeClostridium histolyticumcollagenase, are already used in enzymatic wound debridement and treating fibrotic diseases [84,85].
Lastly, nattokinase fromBacillus subtilisexhibits fibrinolytic and anti-inflammatory activities [86,87]. Directed evolution and DNA family shuffling produced mutants with up to 2.3-fold increased catalytic efficiency [88]. In a separate study, the acidic stability of nattokinase was enhanced through pH-induced buffering and mutagenesis, identifying mutations that improved acid resistance and extended half-life by three-fold, though with slightly reduced activity [89].
Protease-mediated degradation of amyloidogenic proteins
Another area where proteases could unearth new opportunities and fulfill critical needs is in the degradation of aggregated proteins in amyloidosis. Amyloidosis is associated with a broad spectrum of diseases, including AD and Parkinson’s disease (PD), in which amyloid fibrils—insoluble, β-sheet-rich protein aggregates—accumulate, causing cardiomyopathy, neuropathy, and progressive neurodegeneration [90–92]. These fibrils resist natural clearance, and current treatments primarily slow progression and manage symptoms rather than removing the aggregates [93–95]. Antibody-mediated clearance of protein aggregates, while promising in preclinical settings, has had limited success in patients [96]. The dense packing of β-sheets within Aβ fibrils limits the accessibility of canonical protease cleavage sites, impeding proteolysis; however, cleavage near the termini has been shown to significantly destabilize the fibril structure [8,97–100]. This recognition has spurred interest in targeted proteolysis as a strategy to dismantle pathogenic aggregates (Figure 3).
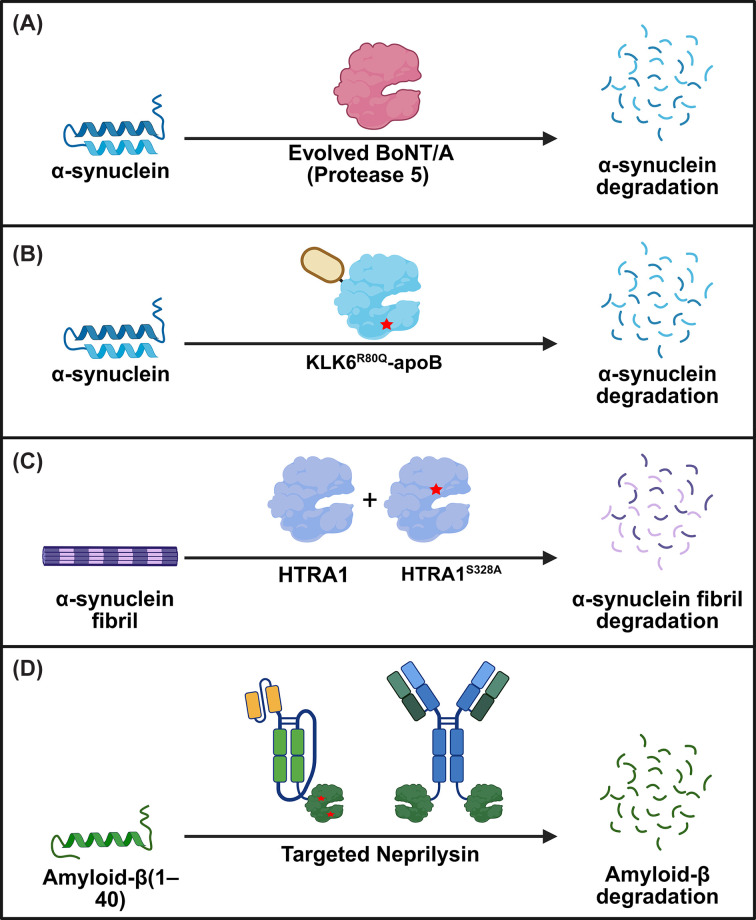
Strategies for the proteolytic degradation of amyloidogenic proteins.(A) A BoNT/A variant (Protease 5), generated through structure-guided mutagenesis, exhibits high catalytic activity and selectivity for α-synuclein (α-syn). (B) The highly stable KLK6R80Q variant is fused to a brain-targeting tag (apoB) and effectively reduces α-syn accumulation. (C) Pre-treatment of α-syn fibrils with the inactive HTRA1 variant, HTRA1S328A, exposes previously inaccessible epitopes, allowing active HTRA1 to cleave and degrade pre-formed fibrils. (D) Several targeted-Neprilysin formats, including scFv-NEP and antibody-NEP, guide catalytic activity to degrade amyloid-β. Abbreviations: KLK6, kallikrein-6; ApoB, apolipoprotein B; HTRA1, high-temperature requirement serine protease A1; BBB, blood–brain barrier.
Proteolytic degradation of misfolded α-synuclein as a possible treatment avenue against synucleopathies
Synucleinopathies are a class of neurodegenerative disorders characterized by the accumulation of α-syn in neurons and glia [101]. When dysregulated, α-syn aggregates spread in a prion-like manner, disrupting neuronal synaptic vesicle trafficking and promoting soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex assembly [102,103]. Targeted degradation of α-syn is therefore an attractive therapeutic goal.
One promising avenue is the reprogramming of proteases to selectively cleave α-syn. A notable example is the catalytic light chain of botulinum neurotoxin A (BoNT/A), a zinc metalloprotease with exceptional sequence specificity for its natural substrate SNAP25. Despite minimal homology between SNAP25 and α-syn, BoNT/A’s extended (>30 amino acid) substrate-binding interface and structurally distinct catalytic and substrate-recognition sites were leveraged to identify an α-syn target site in the non-amyloid core (NAC) region between Q79 and K80 [104]. Through a stepwise, structure-guided mutagenesis campaign spanning 45 sites, a novel BoNT/A variant (Protease 5) was generated with high catalytic activity and selectivity for α-syn. Protease 5 efficiently depleted overexpressed α-syn in mammalian cells while maintaining minimal off-target activity. Importantly, this work establishes a broadly adaptable framework for reprogramming BoNT/A to target other intrinsically disordered proteins implicated in disease.
Another approach for clearing extracellular α-syn aggregates involves the serine protease kallikrein-6 (KLK6, neurosin), which endogenously degrades α-syn. However, KLK6 has many physiological substrates, including protease-activated receptors and glutamate receptors [105]. This unoptimized substrate specificity raises off-target concerns [106,107]. Engineering KLK6 for improved stability and α-syn selectivity has shown therapeutic potential. For example, an R80Q variant with extended half-life and an apolipoprotein B (apoB) brain-targeting tag reduced α-syn accumulation in oligodendrocytes and astrocytes, improved myelination, and ameliorated behavioral deficits in a multiple system atrophy mouse model [108]. Whether KLK6 can be evolved for dramatically improved α-syn potency and selectivity remains to be determined.
HTRA1, a trimeric enzyme with N-terminal, protease, and PDZ domains, can proteolyze and inhibit the aggregation of multiple aggregation-prone proteins, including α-syn, tau, TDP-43, and FUS [97,109]. This chaperone-like “disaggregase” activity is independent of ATP and, surprisingly, can be enhanced by catalytic inactivation (HTRA1S328), which more effectively suppresses α-syn seeding in cells and neurons [97]. HTRA1 engages the α-syn NAC domain within fibrils, solubilizing it and exposing protease-accessible sites. Although it also acts on other amyloidogenic proteins, its potency is greatest for α-syn, and it does not target well-folded proteins. Given its broad substrate range, therapeutic strategies may favor protease-inactive or engineered variants to selectively promote disaggregation while avoiding the degradation of native proteins, making HTRA1 a promising starting point for engineering therapeutics to treat amyloid-related diseases.
Antibody-guided neprilysin for targeted amyloid-β degradation
Accumulation of amyloid-β (Aβ) species and plaques is considered to be a driver of AD pathogenesis [110]. An effective protease-based therapeutic against Aβ would need to cross the blood–brain barrier (BBB), achieve sufficient brain retention, and cleave only the target protein. Two different efforts using Neprilysin (NEP) underscore these strategies. NEP, a membrane-bound metallopeptidase, degrades Aβ but has broad substrate specificity that raises concerns about off-target effects [111,112]. Engineering efforts yielded muNEP (G399V/G714K), which showed 20-fold higher activity on Aβ(1–40) and a 3200-fold reduction in off-target cleavage (assessed across a panel of 16 known physiological peptides, including insulin, glucagon, and neurokinins) relative to wild-type NEP [113]. Despite these improvements, intravenous administration of NEP showed no effect on brain Aβ levels, underscoring the need for better transport through the BBB and improved retention at the target site [113].
To enhance NEP's ability to cross the BBB, researchers fused a transferrin receptor-binding moiety (scFv8D3) to a soluble form of NEP (sNEP) and muNEP, along with an Fc-based antibody fragment (scFc) to extend blood half-life [114]. These fusions achieved a prolonged blood half-life and approximately 20-fold greater brain uptake compared with soluble NEP alone. While the engineered proteins reduced monomeric and oligomeric Aβ levels in tg-ArcSwe mice, brain concentrations declined rapidly, indicating limited retention. Notably, the muNEP fusion failed to degrade wild-type-Aβ at higher Aβ concentrations (2.5 μM). This is likely this is because Aβ concentration was high enough for aggregation to occur, and muNEP has a strong preference for cleaving Aβ at position 20-21, a site that becomes inaccessible in Aβ aggregates. Together, these findings demonstrate that while antibody-guided BBB transport can substantially improve brain delivery of proteases, there is still a need for improved retention.
In an effort to increase the specificity of NEP for Aβ Romei and colleagues fused NEP to crenezumab, an Aβ-specific antibody, producing a C-terminal fusion with 15-fold higher potency for Aβ (1–40) and 9-fold higher potency for Aβ(1–42) compared with untargeted NEP [8]. Interestingly, faster antibody off-rates correlated with greater enzymatic cleavage, suggesting that optimizing binding kinetics could further boost efficacy. While promising,in vivoBBB penetration for such fusions remains untested. More broadly, this study illustrates how antibody–protease fusions can address key limitations of proteases as therapeutics by conferring increased target specificity while preserving the catalytic, sub-stoichiometric turnover that distinguishes enzymes from binding-based modalities.
More broadly, fusion of proteases to targeting domains such as antibodies or nanobodies represents a powerful strategy to enhance substrate specificity and localize catalytic activity. Nb–protease fusions, in particular, offer several advantages, including small size, high stability, and ease of genetic fusion [115]. However, their development introduces several engineering challenges. Fusion can impose steric constraints that reduce catalytic efficiency or limit substrate access, necessitating careful optimization of linker length, flexibility, and fusion orientation [116]. In addition, maintaining the structural stability of both the protease and targeting domain is critical, as misfolding or aggregation can compromise activity and manufacturability. From a production standpoint, while nanobodies are generally amenable to high-yield expression in microbial systems, the inclusion of protease domains and post-translational requirements may necessitate more complex expression platforms, increasing the cost of goods relative to simpler biologics. Despite these challenges, advances in protein engineering and expression technologies are steadily improving the scalability and clinical feasibility of these multifunctional constructs.
Protease-mediated inhibition of upper respiratory viral propagation
ColdZyme is a medical device mouth spray developed by Enzymatica AB, designed to reduce viral propagation in the upper respiratory tract by protecting airway epithelial integrity and limiting viral release from infected cells [117]. Most upper respiratory tract infections (URTIs) are initiated in the mucus of the nasopharynx before spreading throughout the nasal cavity, suggesting that early inhibition of viral propagation in the pharyngeal region during the incubation phase may shorten UTRI duration [118]. ColdZyme consists of a hyperosmotic glycerol solution containing a cold-adapted trypsin derived from Atlantic cod (Gadus morhua), which forms a temporary barrier when sprayed onto the back of the mouth. The proposed mechanism is that viral particles become physically trapped within this barrier and are subsequently inactivated, in part through trypsin's cleavage of viral surface proteins required for receptor binding and host cell entry [119]. This mechanism has not yet been proven experimentally.
In vitrovirucidal assays demonstrate that ColdZyme inactivates multiple respiratory viruses, including rhinovirus types 1A and 42, influenza A virus (H3N2), respiratory syncytial virus, adenovirus type 2, and coronaviruses [119,120]. Early clinical evaluation included a 5-month open-label study in elderly care facility personnel, which reported a reduction in sick-leave days from 5.2 to 3.7 days (29%) during the ColdZyme use period compared with a control period (P= 0.054), with 63% of participants reporting milder symptoms than during previous colds [121]. More recent studies have examined ColdZyme efficacy under free-living conditions in endurance athletes, a population at elevated risk of URTI. In a 3-month randomized trial comparing ColdZyme to no treatment, illness incidence did not differ between groups; however, URTI episode duration was significantly shorter with ColdZyme (7.7 ± 4.0 versus 10.4 ± 8.5 days;P= 0.016) [118]. A subsequent randomized, double-blind, placebo-controlled trial in active athletes further demonstrated reductions in symptom duration (mean reduction ∼5 days), missed training days (mean reduction ∼2.4 days), and symptom severity [117]. Importantly, pathogen-specific analyses indicated that rhinovirus load was markedly reduced by ColdZyme treatment, whereasHaemophilus influenzaewas less responsive, suggesting differential efficacy depending on the causative agent. Consistent with these clinical findings,in vitrostudies using differentiated primary human airway epithelial cultures showed that a single prophylactic application of ColdZyme significantly reduced apical and basolateral release of two non-enveloped rhinovirus strains (RV35 and RV48), preserved transepithelial electrical resistance, and protected epithelial architecture from virus-induced damage. Importantly, there was no difference between groups on URTI incidence, even when subjects used the product preventatively.
Collectively, these data indicate that ColdZyme does not prevent URTI acquisition but instead limits local viral propagation and epithelial damage, resulting in reduced symptom severity and illness duration, particularly in rhinovirus-driven infections. Future work could examine whether efficacy can be further improved by replacing the cod-derived trypsin with an engineered protease optimized to cleave conserved viral surface proteins involved in host cell entry.
Conclusion and future directions
Across immune modulation, infectious disease, metabolic disorders, and neurodegeneration, proteases are rapidly emerging as a versatile therapeutic class. The sections of the present review highlight a consistent theme: despite diverse origins and mechanisms, therapeutic proteases face common barriers such as immunogenicity, limited half-life, insufficient specificity, and challenges in targeted delivery that have historically constrained their clinical use. These limitations have led to proteases being viewed as niche or high-risk agents relative to antibodies and small molecules. However, this perception is becoming increasingly outdated.
Immunomodulatory proteases, such as IgG- and IgM-cleaving enzymes, show strong clinical potential but require innovations that mitigate immunogenicity, preserve protective antibodies, and enable repeat dosing. To facilitate broader clinical application, engineering strategies including humanization, de-immunization, glycosylation, and zymogen design are being developed to improve enzyme stability, reduce immunogenicity, and enable repeated dosing. Successfully addressing these challenges will be crucial to unlocking the full potential of protease-based immune modulation across autoimmune diseases, transplantation, gene therapy, and beyond.
Microbial and plant proteases exhibit broad antiviral activity, metabolic modulation, and toxin degradation. Yet translating these enzymes into medicines demands improved substrate specificity, enhanced stability, and formulations that preserve activityin vivo. Advances in metagenomics, directed evolution, and stability engineering promise to unlock far more of this molecular diversity for therapeutic use.
Proteases targeting misfolded or aggregated proteins illustrate the unique power of enzymes to remodel pathogenic structures rather than merely manage symptoms. Key challenges remain—particularly precision, delivery across the blood-brain barrier, and avoidance of off-target degradation—but the field is advancing toward multifunctional, highly selective proteases guided by nanobodies, engineered domains, or conditional activation mechanisms.
The ability to reprogram proteases for precise therapeutic applications represents a transformative approach in protein engineering. By integrating directed evolution, substrate specificity tuning, and innovative targeting strategies, researchers have made significant progress in enhancing the clinical utility of proteases. The engineering of BoNT and nattokinase demonstrates how modifications can enhance catalytic properties and stability, while the development of proteases targeting neurodegenerative aggregates underscores the potential to treat protein misfolding disorders.
Despite these advances, significant hurdles remain. One of the key challenges is achieving targeted activation, i.e., developing proteases that remain inactive until they reach their intended substrate or location, thereby minimizing off-target effects. Inspired by systems like CRISPR–Cas, in which enzymatic activity is triggered by specific binding events, future efforts may focus on designing allosterically controlled proteases that activate only upon binding their pathological target. This approach could dramatically enhance therapeutic specificity while preserving normal physiological functions.
Beyond intrinsic control strategies, Nb-guided protease modulation represents a rapidly emerging approach for extrinsic regulation of protease activity and substrate selectivity. By binding to noncatalytic or regulatory regions, Nbs can fine-tune protease function rather than abolish it, offering a level of precision difficult to achieve with traditional inhibitors. For example, activated protein C, llama-derived Nbs were identified that selectively recognize the active protease over its zymogen and bias its multifunctional activity toward cytoprotective signaling while preserving coagulation balance [122]. Similarly, furin-specific Nbs isolated via phage panning bind distal domains such as the P-domain, sterically restricting access of large pathogenic substrates like diphtheria toxin without broadly suppressing proteolytic activity [123,124].
A particularly important limitation for protease therapeutics is restricted access to intracellular targets and the central nervous system. Most proteases are large, hydrophilic macromolecules that lack intrinsic membrane permeability, confining their activity primarily to extracellular substrates [125]. Similarly, the BBB presents a formidable obstacle, preventing passive diffusion of proteases into the brain. Consequently, the successful deployment of therapeutic proteases requires engineering delivery strategies alongside catalytic function. Strategies to overcome these barriers include receptor-mediated transcytosis, nanoparticle-based delivery systems, and fusion to BBB-shuttling domains [126]. Notably, an inactivated botulinum neurotoxin (BoNT) platform has been repurposed for Nb-guided, cell-type-specific cytosolic delivery of protein cargos by replacing its native receptor-binding domain with nanobodies directed against selected surface markers [127]. By Retaining BoNT's efficient endosomal escape machinery while eliminating toxicity, this system demonstrates how toxin-derived scaffolds can be repurposed for precise and programmable intracellular delivery of therapeutic proteases.
As synthetic biology, computational protein design, and high-throughput screening continue to converge, the future of protease therapeutics lies in creating enzymes that are not only potent and specific but also programmable and regulatable. Such innovations promise to redefine therapeutic paradigms across neurodegeneration, thrombosis, autoimmune disease, and beyond.
References
- BleuezC. , KochW. F. , UrbachC. , HollfelderF. andJermutusL. (2022)Exploitingprotease activation for therapy. Drug Discov. Today27, 1743–1754 doi.org/10.1016/j.drudis.2022.03.011
- CraikC. S. , PageM. J. andMadisonE. L. (2011)Proteases as therapeutics. Biochem. J. 435, 1–16 doi.org/10.1042/BJ20100965
- BondJ. S. (2019)Proteases: history, discovery, and roles in health and disease. J. Biol. Chem. 294, 1643–1651 doi.org/10.1074/jbc.TM118.004156
- DeraisonC. andVergnolleN. (2026)Proteases in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 23, 6–28 doi.org/10.1038/s41575-025-01129-w
- KangD. andCraikC. S. (2025)Identifying protease-activated targets and exploring therapeutic applications. Expert Opin. Ther. Targets29, 687–701 doi.org/10.1080/14728222.2025.2563244
- MustafaM. , AhmadR. , TantryI. Q. , AhmadW. , SiddiquiS. , AlamM. et al. (2024)Apoptosis: a comprehensive overview of signaling pathways, morphological changes, and physiological significance and therapeutic implications. Cells13, 1838 doi.org/10.3390/cells13221838
- GoldbergK. , LobovA. , AntonelloP. , ShmueliM. D. , YakirI. , WeizmanT. et al. (2025)Cell-autonomous innate immunity by proteasome-derived defence peptides. Nature639, 1032–1041 doi.org/10.1038/s41586-025-08615-w
- RomeiM. G. , LeonardB. , KimI. , KimH. S. andLazarG. A. (2023)Antibody-guided proteases enable selective and catalytic degradation of challenging therapeutic targets. J. Biol. Chem. 299, 104685 doi.org/10.1016/j.jbc.2023.104685
- MoronitiJ. J. , VrbenskyJ. R. , NazyI. andArnoldD. M. (2024)Targeted ADAMTS-13 replacement therapy for thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 22, 896–904 doi.org/10.1016/j.jtha.2023.11.030
- LiuM. , SvirskisD. , ProftT. , LohJ. , YinN. , LiH. et al. (2025)Progress in peptide and protein therapeutics: challenges and strategies. Acta. Pharm. Sin. B. 15, 6342–6381 doi.org/10.1016/j.apsb.2025.10.026
- Cerf-BensussanN. , Matysiak-BudnikT. , CellierC. andHeymanM. (2007)Oral proteases: a new approach to managing coeliac disease. Gut56, 157–160 doi.org/10.1136/gut.2005.090498
- TripathiN. K. andShrivastavaA. (2019)Recent developments in bioprocessing of recombinant proteins: expression hosts and process development. Front. Bioeng. Biotechnol. 7, 420 doi.org/10.3389/fbioe.2019.00420
- LawrenceP. B. andPriceJ. L. (2016)How PEGylation influences protein conformational stability. Curr. Opin. Chem. Biol. 34, 88–94 doi.org/10.1016/j.cbpa.2016.08.006
- KaramitrosC. S. , SomodyC. M. , AgnelloG. andRowlinsonS. (2021)Engineering of the recombinant expression and PEGylation efficiency of the therapeutic enzyme human thymidine phosphorylase. Front. Bioeng. Biotechnol. 9, 793985 doi.org/10.3389/fbioe.2021.793985
- DaventureV. , Bou-JaoudehM. , HannachiE. , Reyes-RuizA. , TreccoA. , DelignatS. et al. (2025)Half-life extension of the IgG-degrading enzyme (IdeS) using Fc-fusion technology. Eur. J. Immunol. 55, e202451264 doi.org/10.1002/eji.202451264
- DyerR. P. andWeissG. A. (2022)Making the cut with protease engineering. Cell Chem. Biol. 29, 177–190 doi.org/10.1016/j.chembiol.2021.12.001
- NelsonS. , GazaJ. , AjayebiS. , MasseR. , PhoR. , ScuteroC. et al. (2025)PERRC: protease engineering with reactant residence time control. ACS Synth. Biol. 14, 2241–2253 doi.org/10.1021/acssynbio.5c00154
- MartinusenS. G. , NelsonS. E. , SlatonE. W. , LongL. F. , PhoR. , AjayebiS. et al. (2025)Protease engineering: approaches, tools, and emerging trends. Biotechnol. Adv. 82, 108602 doi.org/10.1016/j.biotechadv.2025.108602
- HuberL. , KuceraT. , HöllererS. , BorgwardtK. , PankeS. andJeschekM. (2025)Data-driven protease engineering by DNA-recording and epistasis-aware machine learning. Nat. Commun. 16, 5466 doi.org/10.1038/s41467-025-60622-7
- LiQ. , YiL. , MarekP. andIversonB. L. (2013)Commercial proteases: present and future. FEBS Lett. 587, 1155–1163 doi.org/10.1016/j.febslet.2012.12.019
- KhanM. F. andKhanM. T. (2025)AI-driven enzyme engineering: emerging models and next-generation biotechnological applications. Molecules31, 45 doi.org/10.3390/molecules31010045
- AhernW. , YimJ. , TischerD. , SalikeS. , WoodburyS. M. , KimD. et al. (2026)Atom-level enzyme active site scaffolding using RFdiffusion2. Nat. Methods23, 96–105 doi.org/10.1038/s41592-025-02975-x
- MuczynskiV. , CasariC. , MoreauF. , AyméG. , KaweckiC. , LegendreP. et al. (2018)A factor VIII–nanobody fusion protein forming an ultrastable complex with VWF: effect on clearance and antibody formation. Blood132, 1193–1197 doi.org/10.1182/blood-2018-01-829523
- GelbeneggerG. , SchoergenhoferC. , KnoeblP. andJilmaB. (2020)Bridging the missing link with emicizumab: a bispecific antibody for treatment of hemophilia A. Thromb. Haemost. 120, 1357–1370 doi.org/10.1055/s-0040-1714279
- HermansC. andPierceG. F. (2023)Bispecific antibodies mimicking factor VIII in hemophilia A: converting innovation to an essential medicine. Res. Pract. Thromb. Haemost. 7, 100173 doi.org/10.1016/j.rpth.2023.100173
- JiangD. , WangM. , WheelerA. P. andCroteauS. E. (2025)2025 clinical trials update on hemophilia, VWD, and rare inherited bleeding disorders. Am. J. Hematol. 100, 666–684 doi.org/10.1002/ajh.27602
- FarajA. , KnudsenT. , DesaiS. , NeumanL. , BlouseG. E. andSimonssonU. S. H. (2022)Phase III dose selection of marzeptacog alfa (activated) informed by population pharmacokinetic modeling: a novel hemostatic drug. CPT Pharmacometrics Syst. Pharmacol. 11, 1628–1637 doi.org/10.1002/psp4.12872
- MahlanguJ. , LevyH. , KosinovaM. V. , KhachatryanH. , KorczowskiB. , MakhaldianiL. et al. (2021)Subcutaneous engineered factor VIIa marzeptacog alfa (activated) in hemophilia with inhibitors: Phase 2 trial of pharmacokinetics, pharmacodynamics, efficacy, and safety. Res. Pract. Thromb. Haemost. 5, e12576 doi.org/10.1002/rth2.12576
- GruppoR. A. , MalanD. , KapocsiJ. , NemesL. , HayC. R. M. , BoggioL. et al. (2018)Phase 1, single‐dose escalating study of marzeptacog alfa (activated), a recombinant factor VIIa variant, in patients with severe hemophilia. J. Thromb. Haemost. 16, 1984–1993 doi.org/10.1111/jth.14247
- CroteauS. E. , WangM. andWheelerA. P. (2021)2021 clinical trials update: innovations in hemophilia therapy. Am. J. Hematol. 96, 128–144 doi.org/10.1002/ajh.26018
- AnaK. (2021)Catalyst biosciences announces change in corporate strategy. Gyre Therapeutics. Last accessed 11 May 2026
- GC Biopharma USA(2023)GC Biopharma Signs Agreement with Catalyst Biosciences for the Acquisition of Rare Disease Pipeline in Hematology. Available from: Last accessed on May 11th 2026
- Catalyst Biosciences Inc(2021)Protease medicines to catalyse change in complement-targeting therapies. Nature. Last accessed 11 May 2026
- ColtonT. (2022)Catalyst biosciences sells complement portfolio for $60 Million. Gyre Therapeutics. Last accessed 11 May 2026
- PisetskyD. S. (2023)Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 19, 509–524 doi.org/10.1038/s41581-023-00720-1
- BoesM. , SchmidtT. , LinkemannK. , BeaudetteB. C. , Marshak-RothsteinA. andChenJ. (2000)Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. U. S. A. 97, 1184–1189 doi.org/10.1073/pnas.97.3.1184
- HuangH. (2023)Immunotherapeutic approaches for systemic lupus erythematosus: early overview and future potential. Med. Rev. 3, 452–464 doi.org/10.1515/mr-2023-0032
- Guerreiro CastroS. andIsenbergD. A. (2017)Belimumab in systemic lupus erythematosus (SLE): evidence-to-date and clinical usefulness. Ther. Adv. Musculoskelet. 9, 75–85 doi.org/10.1177/1759720X17690474
- SrivastavaA. (2016)Belimumab in systemic lupus erythematosus. Indian J. Dermatol. 61, 550–553 doi.org/10.4103/0019-5154.190107
- SchroederH. W. andCavaciniL. (2010)Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 125, S41–S52 doi.org/10.1016/j.jaci.2009.09.046
- BrezskiR. J. andJordanR. E. (2010)Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity?mAbs2, 212–220 doi.org/10.4161/mabs.2.3.11780
- FrickI. M. , HapponenL. , WrightonS. , NordenfeltP. andBjörckL. (2023)IdeS, a secreted proteinase ofStreptococcus pyogenes, is bound to a nuclease at the bacterial surface where it inactivates opsonizing IgG antibodies. J. Biol. Chem. 299, 105345 doi.org/10.1016/j.jbc.2023.105345
- JordanS. C. , LorantT. , ChoiJ. , KjellmanC. , WinstedtL. , BengtssonM. et al. (2017)IgG endopeptidase in highly sensitized patients undergoing transplantation. N. Engl. J. Med. 377, 442–453 doi.org/10.1056/NEJMoa1612567
- KanbayM. , CopurS. , GuldanM. , TopcuA. U. , OzbekL. , HasbalB. et al. (2024)Imlifidase in kidney transplantation. Clin. Kidney J. 17, sfae033 doi.org/10.1093/ckj/sfae033
- DeLauraI. , ZikosJ. , AnwarI. J. , YoonJ. , LadowskiJ. , JacksonA. et al. (2024)The impact of IdeS (imlifidase) on allo-specific, xeno-reactive, and protective antibodies in a sensitized rhesus macaque model. Xenotransplantation31, e12833 doi.org/10.1111/xen.12833
- WinstedtL. , JärnumS. , NordahlE. A. , OlssonA. , RunströmA. , BockermannR. et al. (2015)Complete removal of extracellular IgG antibodies in a randomized dose-escalation phase I study with the bacterial enzyme IdeS—a novel therapeutic opportunity. PloS One10, e0132011 doi.org/10.1371/journal.pone.0132011
- RathT. , BakerK. , DumontJ. A. , PetersR. T. , JiangH. , QiaoS. -W. et al. (2015)Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 35, 235–254 doi.org/10.3109/07388551.2013.834293
- RostaingL. , NobleJ. , MalvezziP. andJouveT. (2023)Imlifidase therapy: exploring its clinical uses. Expert Opin. Pharmacother. 24, 259–265 doi.org/10.1080/14656566.2022.2150965
- LorantT. , LonzeB. E. , MontgomeryR. A. , DesaiN. M. , LegendreC. , LundgrenT. et al. (2025)Five years follow-up of imlifidase desensitized kidney transplant recipients. Transpl. Int. 38, 15425 doi.org/10.3389/ti.2025.15425
- Hansa Biopharma AB(2025)Imlifidase successfully meets primary endpoint in pivotal US Phase 3 ConfIdeS trial in kidney transplantation. Available from: . Last accessed on May 11th 2026
- Hansa Biopharma AB(2025)Hansa provides update on Pivotal Phase 3 trial in anti-glomerular basement membrane (anti-GBM) disease. Available from: . Last accessed on May 11th 2026
- Hansa Biopharma AB(2024)Hansa Biopharma’s HNSA-5487 achieved rapid and highly robust IgG reduction by more than 95% and clear redosing potential in first-in-human trial. Available from: Last accessed on May 11th 2026
- ChenM. , KimB. , JarvisM. I. , FleuryS. , DengS. , NouraeinS. et al. (2023)Immune profiling of adeno-associated virus response identifies B cell-specific targets that enable vector re-administration in mice. Gene Ther. 30, 429–442 doi.org/10.1038/s41434-022-00371-0
- WangD. , TaiP. W. L. andGaoG. (2019)Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378 doi.org/10.1038/s41573-019-0012-9
- MingozziF. , AnguelaX. M. , PavaniG. , ChenY. , DavidsonR. J. , HuiD. J. et al. (2013)Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 5, 194ra92 doi.org/10.1126/scitranslmed.3005795
- LeborgneC. , BarbonE. , AlexanderJ. M. , HanbyH. , DelignatS. , CohenD. M. et al. (2020)IgG-cleaving endopeptidase enablesin vivogene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 26, 1096–1101 doi.org/10.1038/s41591-020-0911-7
- WangJ. -H. , GesslerD. J. , ZhanW. , GallagherT. L. andGaoG. (2024)Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal. Transduct. Target. Ther. 9, 78 doi.org/10.1038/s41392-024-01780-w
- SmithT. J. , ElmoreZ. C. , FuscoR. M. , HullJ. A. , RosalesA. , MartinezM. et al. (2024)Engineered IgM and IgG cleaving enzymes for mitigating antibody neutralization and complement activation in AAV gene transfer. Mol. Ther. 32, 2080–2093 doi.org/10.1016/j.ymthe.2024.05.004
- BayerA. C. , SanmarcoL. M. , PellerinA. , MasiG. , PlasenciaA. , AndersonJ. M. et al. (2025)Therapeutic IgG- and IgM-specific proteases disarm the acetylcholine receptor autoantibodies that drive myasthenia gravis pathology. Proc. Natl. Acad. Sci. U. S. A. 122, e2505984122 doi.org/10.1073/pnas.2505984122
- GrönwallC. , VasJ. andSilvermanG. J. (2012)Protective roles of natural IgM antibodies. Front. Immunol. 3, 66 doi.org/10.3389/fimmu.2012.00066
- Seismic Therapeutic(2025)Seismic Therapeutic Doses First Cohort in Phase 1 Clinical Trial of S-1117, a Novel Pan IgG Protease Therapy for Antibody-Mediated Diseases. Available from: Last accessed 11 May 2026
- ManassonJ. , SanmarcoL. , PellerinA. , AndersonJ. , RollinsN. , GreenT. et al. (2024)Preclinical polypharmacology of S-1117, a novel engineered Fc-fused IgG degrading enzyme, for chronic treatment of autoantibody-mediated diseases. Blood144, 2562 doi.org/10.1182/blood-2024-201798
- Seismic Therapeutic(2025)Seismic Therapeutic Presents Preclinical Data on S-8484, Its Novel IgE Protease, for Treatment of Allergic Diseases. Available from:
- BéginP. , WasermanS. , ProtudjerJ. L. P. , JeimyS. andWatsonW. (2024)Immunoglobulin E (IgE)-mediated food allergy. Ann. Allergy, Asthma & Clinical Immunology20, 75 doi.org/10.1186/s13223-024-00930-7
- WangL. , LiX. , ShenH. , MaoN. , WangH. , CuiL. et al. (2016)Bacterial IgA protease-mediated degradation of agIgA1 and agIgA1 immune complexes as a potential therapy for IgA Nephropathy. Sci. Rep. 6, 30964 doi.org/10.1038/srep30964
- ZhaoN. , HouP. , LvJ. , MoldoveanuZ. , LiY. , KirylukK. et al. (2012)The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 82, 790–796 doi.org/10.1038/ki.2012.197
- LechnerS. M. , AbbadL. , BoedecE. , PapistaC. , Le StangM. -B. , MoalC. et al. (2016)IgA1 protease treatment reverses mesangial deposits and hematuria in a model of IgA nephropathy. J. Am. Soc. Nephrol. 27, 2622–2629 doi.org/10.1681/ASN.2015080856
- SpoerryC. , KarlssonJ. , AschtgenM. -S. andLohE. (2021)Neisseria meningitidis IgA1-specific serine protease exhibits novel cleavage activity against IgG3. Virulence12, 389–403 doi.org/10.1080/21505594.2021.1871822
- BartonJ. C. , BertoliL. F. , BartonJ. C. andActonR. T. (2016)Selective subnormal IgG3 in 121 adult index patients with frequent or severe bacterial respiratory tract infections. Cell. Immunol. 299, 50–57 doi.org/10.1016/j.cellimm.2015.09.004
- WeiG. , HelmerhorstE. J. , DarwishG. , BlumenkranzG. andSchuppanD. (2020)Gluten degrading enzymes for treatment of celiac disease. Nutrients12, 2095. doi.org/10.3390/nu12072095
- KönigJ. , HolsterS. , BruinsM. J. andBrummerR. J. (2017)Randomized clinical trial: Effective gluten degradation byAspergillus niger-derived enzyme in a complex meal setting. Sci. Rep. 7, 13100 doi.org/10.1038/s41598-017-13587-7
- HarrisC. T. andCohenS. (2024)Reducing immunogenicity by design: approaches to minimize immunogenicity of monoclonal antibodies. BioDrugs38, 205–226 doi.org/10.1007/s40259-023-00641-2
- CavalettiL. , TaravellaA. , CarranoL. , CarenziG. , SigurtàA. , SolinasN. et al. (2019)E40, a novel microbial protease efficiently detoxifying gluten proteins, for the dietary management of gluten intolerance. Sci. Rep. 9, 13147 doi.org/10.1038/s41598-019-48299-7
- MamoneG. , ComelliM. C. , VitaleS. , Di StasioL. , KesslerK. , MottolaI. et al. (2022)E40 glutenase detoxification capabilities of residual gluten immunogenic peptides inin vitrogastrointestinal digesta of food matrices made of soft and durum wheat. Front. Nutr. 9, 974771 doi.org/10.3389/fnut.2022.974771
- XiaoB. , ZhangC. , ZhouJ. , WangS. , MengH. , WuM. et al. (2023)Design of SC PEP with enhanced stability against pepsin digestion and increased activity by machine learning and structural parameters modeling. Int. J. Biol. Macromol. 250, 125933 doi.org/10.1016/j.ijbiomac.2023.125933
- David TroncosoF. , Alberto SánchezD. andLuján FerreiraM. (2022)Production of plant proteases and new biotechnological applications: an updated review. ChemistryOpen11, e202200017 doi.org/10.1002/open.202200017
- VachherM. , SenA. , KapilaR. andNigamA. (2021)Microbial therapeutic enzymes: a promising area of biopharmaceuticals. Curr. Res. Biotechnol. 3, 195–208 doi.org/10.1016/j.crbiot.2021.05.006
- RungsaengP. , SangvanichP. andKarnchanatatA. (2013)Zingipain, a ginger protease with acetylcholinesterase inhibitory activity. Appl. Biochem. Biotechnol. 170, 934–950 doi.org/10.1007/s12010-013-0243-x
- ZhangJ. , ZhangY. , WangJ. , XiaY. , ZhangJ. andChenL. (2024)Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal. Transduct. Target. Ther. 9, 211 doi.org/10.1038/s41392-024-01911-3
- TagaY. , HayashidaO. , KusubataM. , Ogawa-GotoK. andHattoriS. (2017)Production of a novel wheat gluten hydrolysate containing dipeptidyl peptidase-IV inhibitory tripeptides using ginger protease. Biosci. Biotechnol. Biochem. 81, 1823–1828 doi.org/10.1080/09168451.2017.1345615
- YamamotoY. , MizushigeT. , MoriY. , ShimmuraY. , FukutomiR. , KanamotoR. et al. (2015)Antidepressant-like effect of food-derived pyroglutamyl peptides in mice. Neuropeptides51, 25–29 doi.org/10.1016/j.npep.2015.04.002
- YaoH. , LiuS. , LiuT. , RenD. , ZhouZ. , YangQ. et al. (2023)Microbial-derived salt-tolerant proteases and their applications in high-salt traditional soybean fermented foods: a review. Bioresour. Bioprocess. 10, 82 doi.org/10.1186/s40643-023-00704-w
- ChristopherM. , Kooloth-ValappilP. , Sreeja-RajuA. andSukumaranR. K. (2021)Repurposing proteases: anin-silicoanalysis of the binding potential of extracellular fungal proteases with selected viral proteins. Bioresour. Technol. Rep. 15, 100756 doi.org/10.1016/j.biteb.2021.100756
- Mousavi GhahfarrokhiS. S. , MahdigholiF. S. andAminM. (2023)Collateral beauty in the damages: an overview of cosmetics and therapeutic applications of microbial proteases. Arch. Microbiol. 205, 375 doi.org/10.1007/s00203-023-03713-7
- PhamC. H. , CollierZ. J. , FangM. , HowellA. andGillenwaterT. J. (2019)The role of collagenase ointment in acute burns: a systematic review and meta-analysis. J. Wound Care28, S9–S15, doi.org/10.12968/jowc.2019.28.Sup2.S9
- WengY. , YaoJ. , SparksS. andWangK. Y. (2017)Nattokinase: an oral antithrombotic agent for the prevention of cardiovascular disease. Int. J. Mol. Sci. 18, doi.org/10.3390/ijms18030523
- WuH. , WangY. , ZhangY. , XuF. , ChenJ. , DuanL. et al. (2020)Breaking the vicious loop between inflammation, oxidative stress and coagulation, a novel anti-thrombus insight of nattokinase by inhibiting LPS-induced inflammation and oxidative stress. Redox Biol. 32, 101500 doi.org/10.1016/j.redox.2020.101500
- YongjunC. , WeiB. , ShujunJ. , MeizhiW. , YanJ. , YanY. et al. (2011)Directed evolution improves the fibrinolytic activity of nattokinase from Bacillus natto. FEMS Microbiol. Lett. 325, 155–161 doi.org/10.1111/j.1574-6968.2011.02423.x
- LiuZ. , ZhaoH. , HanL. , CuiW. , ZhouL. andZhouZ. (2019)Improvement of the acid resistance, catalytic efficiency, and thermostability of nattokinase by multisite-directed mutagenesis. Biotechnol. Bioeng. 116, 1833–1843 doi.org/10.1002/bit.26983
- BustamanteJ. G. andZaidiS. R. H. (2025)Amyloidosis. InStatPearls, StatPearls Publishing LLC, Treasure Island (FL)
- MetkarS. K. , UdayakumarS. , GirigoswamiA. andGirigoswamiK. (2024)Amyloidosis-history and development, emphasis on insulin and prion amyloids. Brain Disorders13, 100106 doi.org/10.1016/j.dscb.2023.100106
- SuenagaG. , IkedaT. , KomoharaY. , TakamatsuK. , KakumaT. , TasakiM. et al. (2016)Involvement of macrophages in the pathogenesis of familial amyloid polyneuropathy and efficacy of human iPS cell-derived macrophages in its treatment. PloS One11, e0163944 doi.org/10.1371/journal.pone.0163944
- Guerrero-MuñozM. J. , Castillo-CarranzaD. L. andKayedR. (2014)Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochem. Pharmacol. 88, 468–478 doi.org/10.1016/j.bcp.2013.12.023
- CiechanoverA. andKwonY. T. (2015)Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47, e147 doi.org/10.1038/emm.2014.117
- PoeweW. , SeppiK. , TannerC. M. , HallidayG. M. , BrundinP. , VolkmannJ. et al. (2017)Parkinson disease. Nat. Rev. Dis. Primers. 3, 17013 doi.org/10.1038/nrdp.2017.13
- GeertsH. , BergelerS. , WalkerM. , van der GraafP. H. andCouradeJ. -P. (2023)Analysis of clinical failure of anti-tau and anti-synuclein antibodies in neurodegeneration using a quantitative systems pharmacology model. Sci. Rep. 13, 14342 doi.org/10.1038/s41598-023-41382-0
- ChenS. , PuriA. , BellB. , FritscheJ. , PalaciosH. H. , BalchM. et al. (2024)HTRA1 disaggregates α-synuclein amyloid fibrils and converts them into non-toxic and seeding incompetent species. Nat. Commun. 15, 2436 doi.org/10.1038/s41467-024-46538-8
- TatebeH. , WatanabeY. , KasaiT. , MizunoT. , NakagawaM. , TanakaM. et al. (2010)Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci. Res. 67, 341–346 doi.org/10.1016/j.neures.2010.04.008
- LambethT. R. andJulianR. R. (2021)Proteolysis of amyloid β by lysosomal enzymes as a function of fibril morphology. ACS Omega6, 31520–31527 doi.org/10.1021/acsomega.1c03915
- Solé-DomènechS. , RojasA. V. , MaisuradzeG. G. , ScheragaH. A. , LobelP. andMaxfieldF. R. (2018)Lysosomal enzyme tripeptidyl peptidase 1 destabilizes fibrillar Aβ by multiple endoproteolytic cleavages within the β-sheet domain. Proc. Natl. Acad. Sci. U. S. A. 115, 1493–1498 doi.org/10.1073/pnas.1719808115
- ForloniG. (2023)Alpha synuclein: neurodegeneration and inflammation. Int. J. Mol. Sci. 24, 5914 doi.org/10.3390/ijms24065914
- BurréJ. , SharmaM. , TsetsenisT. , BuchmanV. , EthertonM. R. andSüdhofT. C. (2010)α-Synuclein promotes SNARE-complex assemblyin vivoandin vitro. Science329, 1663–1667 doi.org/10.1126/science.1195227
- LautenschlägerJ. , KaminskiC. F. andKaminski SchierleG. S. (2017)α-Synuclein—regulator of exocytosis, endocytosis, or both?Trends Cell Biol. 27, 468–479 doi.org/10.1016/j.tcb.2017.02.002
- SondermannP. , DiercksC. S. , RongC. andSchultzP. G. (2025)Targeted degradation of α-Synuclein using an evolved botulinum toxin protease. Proc. Natl. Acad. Sci. U. S. A. 122, e2426745122 doi.org/10.1073/pnas.2426745122
- LiH. , HwangB. , LaxmikanthanG. , BlaberS. I. , BlaberM. , GolubkovP. A. et al. (2008)Substrate specificity of human kallikreins 1 and 6 determined by phage display. Protein Sci. 17, 664–672 doi.org/10.1110/ps.073333208
- OikonomopoulouK. , HansenK. K. , SaifeddineM. , TeaI. , BlaberM. , BlaberS. I. et al. (2006)Proteinase-activated receptors, targets for kallikrein signaling*. J. Biol. Chem. 281, 32095–32112 doi.org/10.1074/jbc.M513138200
- PampalakisG. , SykiotiV. S. , XimerakisM. , Stefanakou-KalakouI. , MelkiR. , VekrellisK. et al. (2017)KLK6 proteolysis is implicated in the turnover and uptake of extracellular alpha-synuclein species. Oncotarget8, 14502–14515 doi.org/10.18632/oncotarget.13264
- SpencerB. , ValeraE. , RockensteinE. , Trejo-MoralesM. , AdameA. andMasliahE. (2015)A brain-targeted, modified neurosin (kallikrein-6) reduces α-synuclein accumulation in a mouse model of multiple system atrophy. Mol. Neurodegener. 10, 48 doi.org/10.1186/s13024-015-0043-6
- PoepselS. , SprengelA. , SaccaB. , KaschaniF. , KaiserM. , GatsogiannisC. et al. (2015)Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 11, 862–869 doi.org/10.1038/nchembio.1931
- HampelH. , HardyJ. , BlennowK. , ChenC. , PerryG. , KimS. H. et al. (2021)The amyloid-β pathway in Alzheimer’s disease. Mol. Psychiatry26, 5481–5503 doi.org/10.1038/s41380-021-01249-0
- MossS. , SubramanianV. andAcharyaK. R. (2020)Crystal structure of peptide-bound neprilysin reveals key binding interactions. FEBS Lett. 594, 327–336 doi.org/10.1002/1873-3468.13602
- SaxenaS. K. , AnsariS. , MauryaV. K. , KumarS. , SharmaD. , MalhotraH. S. et al. (2024)Neprilysin-mediated amyloid beta clearance and its therapeutic implications in neurodegenerative disorders. ACS Pharmacol. Transl. Sci. 7, 3645–3657 doi.org/10.1021/acsptsci.4c00400
- WebsterC. I. , BurrellM. , OlssonL. -L. , FowlerS. B. , DigbyS. , SandercockA. et al. (2014)Engineering neprilysin activity and specificity to create a novel therapeutic for Alzheimer’s disease. PloS One9, e104001 doi.org/10.1371/journal.pone.0104001
- RofoF. , MetzendorfN. G. , SaubiC. , SuominenL. , GodecA. , SehlinD. et al. (2022)Blood–brain barrier penetrating neprilysin degrades monomeric amyloid-beta in a mouse model of Alzheimer’s disease. Alzheimers Res. Ther. 14, 180 doi.org/10.1186/s13195-022-01132-2
- AlexanderE. andLeongK. W. (2024)Discovery of nanobodies: a comprehensive review of their applications and potential over the past five years. J. Nanobiotechnol. 22, 661 doi.org/10.1186/s12951-024-02900-y
- KuoC. -W. , GökC. , FultonH. , Dickson-MurrayE. , AduS. , GallenE. K. et al. (2025)Nanobody-thioesterase chimeras to specifically target protein palmitoylation. Nat. Commun. 16, 1445 doi.org/10.1038/s41467-025-56716-x
- DavisonG. , SchoemanM. , ChidleyC. , DulsonD. K. , SchweighoferP. , WittingC. et al. (2025)ColdZyme® reduces viral load and upper respiratory tract infection duration and protects airway epithelia from infection with human rhinoviruses. The J. Physiol. 603, 1483–1501 doi.org/10.1113/JP288136
- DavisonG. , PerkinsE. , JonesA. W. , SwartG. M. , JenkinsA. R. , RobinsonH. et al. (2021)Coldzyme® Mouth Spray reduces duration of upper respiratory tract infection symptoms in endurance athletes under free living conditions. Eur. J. Sport Sci. 21, 771–780 doi.org/10.1080/17461391.2020.1771429
- StefanssonB. , GudmundsdottirÁ. andClarsundM. (2017)A medical device forming a protective barrier that deactivates four major common cold viruses. Virol Res. Rev. 1, 1–3 doi.org/10.15761/VRR.1000130
- GudmundsdottirÁ. , SchevingR. , LindbergF. andStefanssonB. (2021)Inactivation of SARS‐CoV‐2 and HCoV‐229Ein vitroby ColdZyme® a medical device mouth spray against the common cold. J. Med. Virol. 93, 1792–1795 doi.org/10.1002/jmv.26554
- ClarsundM. andPerssonC. B. (2017)Evaluation of ColdZyme mouth spray against common cold in elderly care personnel. Open J. Respir. Dis. 7, 12–17 doi.org/10.4236/ojrd.2017.71002
- SimD. S. , ShuklaM. , MallariC. R. , FernándezJ. A. , XuX. , SchneiderD. et al. (2023)Selective modulation of activated protein C activities by a nonactive site–targeting nanobody library. Blood Adv. 7, 3036–3048 doi.org/10.1182/bloodadvances.2022008740
- ZhuJ. , DeclercqJ. , RoucourtB. , GhassabehG. H. , MeulemansS. , KinneJ. et al. (2012)Generation and characterization of non-competitive furin-inhibiting nanobodies. Biochemical J. 448, 73–82 doi.org/10.1042/BJ20120537
- DahmsS. O. , CreemersJ. W. M. , SchaubY. , BourenkovG. P. , ZöggT. , BrandstetterH. et al. (2016)The structure of a furin-antibody complex explains non-competitive inhibition by steric exclusion of substrate conformers. Sci. Rep. 6, 34303 doi.org/10.1038/srep34303
- BaralK. C. andChoiK. Y. (2025)Barriers and strategies for oral peptide and protein therapeutics delivery: update on clinical advances. Pharmaceutics17, 397 doi.org/10.3390/pharmaceutics17040397
- LeiY. , XiaoT. , HuX. andLinH. (2026)Advanced drug delivery strategies for overcoming biological barriers: tumor microenvironment and blood–brain barrier. Drugs R. D. 26, 1–30 doi.org/10.1007/s40268-026-00538-9
- RohH. , DornerB. G. andTingA. Y. (2023)Cell-type-specific intracellular protein delivery with inactivated botulinum neurotoxin. J. Ame. Chem. Soc. 145, 10220–10226 doi.org/10.1021/jacs.3c01145
Republished from the open web under CC-BY. Authors: Nelson SE, Martinusen SG, Pho R, Long LF, Denard CA. Read the original.